Resolución 3496 EXENTA APRUEBA EL INSTRUCTIVO DE FARMACOVIGILANCIA PARA LA ELABORACIÓN DE LOS PLANES DE MANEJO DE RIESGOS ELABORADO POR EL DEPARTAMENTO AGENCIA NACIONAL DE MEDICAMENTOS
MINISTERIO DE SALUD; SUBSECRETARÍA DE SALUD PÚBLICA; INSTITUTO DE SALUD PÚBLICA
Promulgacion: 15-OCT-2013 Publicación: 06-NOV-2013
Versión: Única - 06-NOV-2013
Url: https://www.bcn.cl/leychile/navegar?i=1055899&f=2013-11-06
APRUEBA EL INSTRUCTIVO DE FARMACOVIGILANCIA PARA LA ELABORACIÓN DE LOS PLANES DE MANEJO DE RIESGOS ELABORADO POR EL DEPARTAMENTO AGENCIA NACIONAL DE MEDICAMENTOS
Núm. 3.496 exenta.- Santiago, 15 de octubre de 2013.- Vistos: El Instructivo de Farmacovigilancia para la elaboración de los planes de manejo de riesgos elaborado por el Departamento Agencia Nacional de Medicamentos del Instituto de Salud Pública de Chile;
Teniendo presente: Lo dispuesto en los artículos 60º y 61º letras a), d) y f) del decreto con fuerza de ley 1/2005, que fija el texto refundido, coordinado y sistematizado del decreto ley 2.763, de 1979, y de las leyes 18.933 y 18.469; el decreto Nº 3, del año 2010, del Ministerio de Salud, que aprueba el Reglamento del Sistema Nacional de Control de los Productos Farmacéuticos de Uso Humano; la resolución Nº 1.600, del año 2008, de la Contraloría General de la República; los artículos 8, 10 letra a) del decreto supremo Nº 1.222, de 1996, y considerando lo dispuesto en el decreto Nº 122, del año 2010, del Ministerio de Salud, y
Considerando:
1. Que el Departamento Agencia Nacional de Medicamentos de este Instituto ha elaborado un Instructivo de Farmacovigilancia para la elaboración de los planes de manejo de riesgos, el cual tiene como finalidad contar con una herramienta práctica que facilite la elaboración de los mismos, orientando de manera clara a los titulares de registros sanitarios de productos farmacéuticos acerca de su contenido y el formato que deberá aplicarse.
2. Que la gestión o manejo de riesgos es una parte integral del campo de acción de la Farmacovigilancia, ya que contribuye a la prevención de los riesgos de los medicamentos una vez que ellos han sido comercializados.
3. Que el sistema de gestión de riesgos es un conjunto de actividades e intervenciones destinadas a identificar, caracterizar, prevenir o minimizar los riesgos relacionados con los productos farmacéuticos, así como la evaluación de la efectividad de estas intervenciones.
4. Que el plan de manejo de riesgos es un requisito para determinados productos farmacéuticos, contemplado en la Norma Técnica Nº 140, sobre Sistema Nacional de Farmacovigilancia de Productos Farmacéuticos de Uso Humano.
5. Que es necesario que este instructivo sea aprobado administrativamente para su posterior difusión a la comunidad, es por lo que dicto la siguiente
Resolución:
1. Apruébase el Instructivo de Farmacovigilancia para la elaboración de los planes de manejo de riesgos, cuyo tenor es el siguiente:
GLOSARIO DE CONCEPTOS Y TÉRMINOS
Los siguientes términos se reproducen íntegramente del decreto supremo Nº 3, de 2010, "Reglamento del sistema nacional de control de productos farmacéuticos de uso humano", de la Norma General Técnica Nº 140 sobre sistema nacional de farmacovigilancia de productos farmacéuticos de uso humano y de la resolución exenta Nº 441, de 2012, que establece y actualiza el proceso de notificación de eventos adversos ocurridos en ensayos clínicos que se desarrolla en Chile 1, 2, 3:
Confidencialidad: Mantenimiento de la privacidad de los pacientes, profesionales de la salud e instituciones, incluyendo la identidad de las personas y toda la información médica personal. El ámbito de la confidencialidad en la práctica de FV comprende al paciente, al notificador, al centro asistencial, al ISP y al titular del registro sanitario o cualquiera entidad involucrada en una sospecha de RAM.
Eficacia: Aptitud de un medicamento o producto farmacéutico para producir los efectos terapéuticos propuestos, determinada por métodos científicos y estudios realizados en seres humanos.
Evento adverso: Cualquier incidencia perjudicial para la salud en un paciente o sujeto de ensayo clínico tratado con un medicamento, aunque no tenga necesariamente relación causal con dicho tratamiento.
Folleto de información al profesional: Documento que contendrá a lo menos las características de la especialidad farmacéutica; aspectos farmacocinéticos, farmacodinámicos y toxicológicos del mismo; así como las indicaciones, dosificación, grupo etario al cual va dirigido, contraindicaciones, interacciones, precauciones y/o advertencias, reacciones adversas, dentro de las cuales es preciso señalar las que puedan presentarse durante el embarazo, lactancia o en poblaciones especiales; las medidas a tomar en casos de sobredosis y otros aspectos determinadas por la autoridad en base a la naturaleza e información científica disponible de un producto farmacéutico, con la finalidad de informar a los profesionales legalmente habilitados para prescribir o dispensar productos farmacéuticos.
Folleto de información al paciente: Documento destinado a informar al paciente sobre una especialidad farmacéutica. Contendrá a lo menos la información referente a la indicación autorizada, advertencias, contraindicaciones, interacciones con otros productos, precauciones y toda otra información que la autoridad sanitaria determine en el registro, que permitan asegurar su correcto uso. El folleto de los productos farmacéuticos de venta directa deberá señalar además información acerca de la dosificación habitual.
Monografía: Documento que contiene la descripción técnica, farmacéutica o científica de las características y propiedades de un producto.
Nombre genérico de un producto farmacéutico: Denominación aceptada por la Organización Mundial de la Salud (OMS), bajo los distintivos y siglas "Denominaciones Comunes Internacionales" (DCI) o International Non Proprietary Names" (INN) y en su defecto en las farmacopeas oficialmente reconocidas en el país.
Plan de manejo de riesgos: Documento en el cual el solicitante o titular de un Registro Sanitario especifica los riesgos relevantes del medicamento y establece un plan para la realización de las actividades de FV necesarias a fin de identificarlos, caracterizarlos, cuantificarlos, y, en caso necesario, someterlos a un programa específico de prevención o minimización de dichos riesgos.
Producto farmacéutico o medicamento: Toda sustancia natural o sintética, o mezcla de ellas, que se destine a la administración al ser humano o a los animales, con fines de curación, atenuación, tratamiento, prevención y diagnóstico de las enfermedades o de sus síntomas.
Reacción Adversa al Medicamento (RAM): La reacción nociva y no intencionada que se produce a dosis utilizadas normalmente en el ser humano.
Reacción adversa grave o seria: Cualquier reacción adversa que sea mortal o que pueda poner en peligro la vida o que implique incapacidad o invalidez grave o que tenga por consecuencia la hospitalización o prolongación de la misma.
Registro sanitario: Proceso de evaluación de un producto farmacéutico, que siendo favorable, se traduce en una inscripción en un rol especial con numeración correlativa que mantiene el Instituto, previo a su distribución y uso.
Relación beneficio/riesgo de un medicamento: Es la relación entre el beneficio esperado y el riesgo documentado o esperado que puede derivarse de una intervención terapéutica determinada que involucre un medicamento.
Titular de registro sanitario: Persona natural o jurídica, nacional o extranjera, domiciliada en Chile, a cuyo nombre figura un registro sanitario.
Los funcionarios del Subdepartamento Farmacoviglancia del Dpto. Agencia Nacional de Medicamentos del Instituto de Salud Pública entenderán por beneficio, efectividad, error de medicación, interacción medicamentosa y riesgo, los conceptos que a continuación se detallan, de acuerdo a las definiciones recogidas del Documento Técnico Nº 5 de la Red Panamericana de Armonización de la Reglamentación Farmacéutica, "Buenas Prácticas de Farmacovigilancia para las Américas" de la Organización Panamericana de la Salud, publicado el año 2011 4:
Beneficio (terapéutico): Habitualmente se expresa como el efecto terapéutico demostrado que tiene un producto, aunque también debe incluir la valoración subjetiva del paciente acerca de estos efectos.
Efectividad: Grado en que una determinada intervención origina un resultado beneficioso en las condiciones de la práctica habitual, sobre una población determinada.
Error de medicación: Incidente que ocurre mientras la medicación está bajo control del personal sanitario, el paciente o el consumidor, puede evitarse y es causado por la utilización inadecuada de un medicamento. Puede resultar en daño al paciente.
Interacción medicamentosa: Cualquier interacción entre uno o más medicamentos, entre un medicamento y un alimento, y entre un medicamento y una prueba de laboratorio. Las dos primeras categorías de interacciones tienen importancia por el efecto que producen en la actividad farmacológica del medicamento: aumentan o disminuyen los efectos deseables o los adversos. La importancia de la tercera categoría de interacción reside en la alteración que determinado medicamento puede causar en los resultados de las pruebas de laboratorio afectando su confiabilidad.
Riesgo: Es la probabilidad de ocasionar un perjuicio, que normalmente se expresa como un porcentaje o una razón, la probabilidad de un suceso.
GLOSARIO DE ACRÓNIMOS Y ABREVIATURAS
EA: Evento adverso
FV: Farmacovigilancia
ICH: International Conference on Harmonisation (Conferencia Internacional de Armonización)
IPS: Informe Periódico de Seguridad
OPS (en inglés PAHO, Panamerican Health Organization): Organización Panamericana de la Salud
RAM: Reacción Adversa a Medicamentos
RFV: Responsable de Farmacovigilancia
TRS: Titular del Registro Sanitario
PROCESO DE GESTIÓN DE RIESGOS
En este proceso pueden distinguirse tres etapas interrelacionadas e interactivas entre sí 5:
. Caracterización del perfil de seguridad del producto farmacéutico.
. Planificación de actividades de Farmacovigilancia (FV) para caracterizar los riesgos conocidos e identificar nuevos riesgos, e incrementar el conocimiento en general acerca del perfil de seguridad del producto.
. Planificación e implementación de actividades de minimización y mitigación de riesgos, y la evaluación de la efectividad de estas actividades.
En FV se han establecido tres acciones relevantes de gestión del riesgo 4, 6:
1. Adoptar medidas administrativas de reducción del riesgo:
Estas medidas pueden ser diversas, desde informar del nuevo riesgo hasta la retirada inmediata del producto farmacéutico. Esta decisión siempre deberá estar basada en evidencias, experiencia, objetividad y transparencia y estará a cargo primordialmente de la autoridad reguladora, teniendo siempre en cuenta la aceptabilidad social del riesgo en función del beneficio que entrega el medicamento.
2. Comunicar a los profesionales sanitarios y a los pacientes la existencia del riesgo, las medidas adoptadas y las recomendaciones al respecto:
La comunicación tiene que ver con el derecho tanto de los pacientes como de los profesionales sanitarios de conocer los riesgos a los cuales se podrían ver expuestos. Para ello, es importante definir el grado de información, cómo y cuándo informar, una vez que se haya tomado alguna decisión. Para esto, se debe tener en cuenta que la percepción de riesgo varía de acuerdo a quién se le quiera comunicar; es importante que los pacientes entiendan que la eficacia, y aún la efectividad y riesgo, sólo indican probabilidades de un buen o mal resultado para ellos, con base en información disponible. Por su parte, para los profesionales de la salud, el acceso a la información debe ser expedito y de la forma más completa posible.
3. Establecer estrategias específicas de prevención:
Las estrategias de prevención del riesgo pueden ser variadas; hay que tener en cuenta que las reacciones adversas a medicamentos pueden ser causadas tanto por errores en la fabricación, suministro, prescripción, dispensación o administración del medicamento (reacciones adversas extrínsecas) como por las mismas propiedades farmacológicas inherentes al fármaco (reacciones adversas intrínsecas).
Las reacciones extrínsecas resultan más fáciles de advertir o prevenir, mientras que las intrínsecas no. En efecto, hasta que no se conozca más a fondo el mecanismo que produce dichas reacciones intrínsecas, las actividades de prevención se limitan a medidas legislativas o a restricciones en la prescripción. Dentro de las reacciones extrínsecas las más frecuentes se asocian con errores de medicación, de manera tal que las estrategias para evitar estos errores son de vital importancia.
DISEÑO DEL PLAN DE MANEJO DE RIESGOS
El diseño del plan se analiza caso a caso, de acuerdo al medicamento y teniendo en cuenta 4, 6:
. Su naturaleza y la relación beneficio/riesgo conocida, evaluando:
o Tipo, magnitud y frecuencia de los riesgos y beneficios.
o Poblaciones en mayor riesgo y las que obtendrían mayores beneficios.
o La existencia de tratamientos alternativos.
o La reversibilidad de los eventos adversos observados.
. La prevención de eventos adversos.
. La probabilidad de beneficios.
El desarrollo, implementación y evaluación de un plan de manejo de riesgos forma parte de un esfuerzo de FV para promover un balance beneficio/riesgo satisfactorio, dentro de las condiciones de uso especificadas para el producto farmacéutico 7.
El conocimiento relacionado con el perfil de seguridad del producto farmacéutico puede verse alterado en el tiempo, debido a la extensión de su uso en términos tanto de las características de los pacientes como del número de pacientes expuestos 7.
El balance beneficio/riesgo puede optimizarse mediante la reducción del riesgo o el incremento de los beneficios para los pacientes, a través de una FV efectiva que permita una retroalimentación de información de manera oportuna. De esta manera, el plan de manejo de riesgos forma un ciclo, como se muestra en la Figura Nº 1 5.
Figura Nº 1. Ciclo del Plan de Manejo de Riesgos.
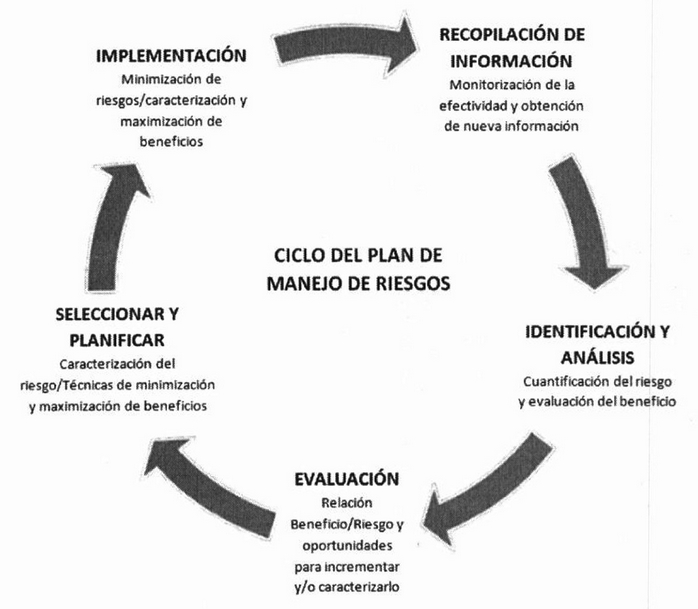
El manejo de riesgos es una actividad global. Sin embargo, debido a que las indicaciones y los sistemas de salud varían en los diferentes lugares del mundo, es posible contar con poblaciones distintas, y las actividades de minimización de riesgo necesitarán adaptarse al sistema particular del país o región. Además, existen diferencias en la prevalencia y severidad de las enfermedades, lo cual podría significar que los beneficios de un producto farmacéutico también varíen entre regiones. De esta manera, un producto farmacéutico puede tener diferentes versiones de planes de manejo de riesgos para cada país o región, aunque los elementos centrales serán comunes en todos 5.
ESTRUCTURA DE UN PLAN DE MANEJO DE RIESGOS
El plan de manejo de riesgos consta de dos partes:
. Parte I. Plan de Farmacovigilancia:
En general, el plan de FV describe en detalle las medidas de FV relacionada a los riesgos potenciales y a los identificados en la especificación de seguridad. Este plan debe describir en detalle las actividades de rutina de FV llevadas a cabo por el titular del registro para el medicamento en particular que se está evaluando 8.
En aquellos productos farmacéuticos para los cuales no han surgido especiales preocupaciones de seguridad, debiera ser suficiente el monitoreo de su seguridad postcomercialización, sin ser necesario actividades adicionales, por lo que un plan de FV sería suficiente. Sin embargo, para productos farmacéuticos con riesgos identificados o riesgo potenciales importantes, o falta de información relevante, deben diseñarse actividades adicionales, las cuales deben ser consideradas dentro de un plan de minimización de riesgos 8.
. Parte II. Plan de Minimización de Riesgos:
El Plan de Minimización de Riesgos es un programa estratégico de seguridad orientado al manejo de nuevos riesgos identificados en el período de postcomercialización o incluso al seguimiento de los riesgos conocidos en poblaciones previamente estudiadas. Es recomendable que este plan se desarrolle con objetivos prácticos, específicos y medibles para poder alcanzar las metas, y esto dependerá del tipo, frecuencia y severidad del riesgo identificado. Actualmente existen diversas estrategias de Minimización de Riesgos 8:
. Educación y alcance: Guías de medicación para pacientes,
programas de educación-continua para proveedores de atención
en salud.
. Sistemas recordatorios: Indicaciones, recordatorios,
dobles chequeos o guías para los proveedores de atención
en salud, pacientes o ambos.
. Educación al paciente con reconocimiento o uso de
formas de consentimiento informado.
. Certificación de capacitación para prestadores de salud.
. Dispensación limitada.
. Desarrollo de sistemas ligados al acceso:
o Prescripción y dispensación únicamente por
profesionales de la salud especialmente entrenados
y certificados.
o Dispensación únicamente bajo condiciones que reúnen
evidencia de requerimientos de uso seguro.
o Inscripción obligatoria o registro de pacientes,
prescriptores o farmacéuticos en programas o
registros de distribución de fármacos restringidos.
o Administración del fármaco en situaciones especiales.
Además de una rutina de FV, los Planes de Minimización de Riesgos deben presentar un propósito basado en métodos farmacoepidemiológicos, donde exista la necesidad de asegurar puntos críticos relacionados a la seguridad del producto farmacéutico 5.
El titular del registro sanitario (TRS) debe justificar el método propuesto para la ejecución de los Planes de Minimización de Riesgos. En este plan, el TRS deberá explicar cómo se evaluará la efectividad de las acciones para minimizar el riesgo asociado a sus productos 8.
MODELO Y ESTRUCTURA PARA LA ELABORACIÓN DE UN PLAN DE MANEJO DE RIESGOS
I. Portada 5, 8
. Número de documento.
. Nombre comercial y genérico del producto.
. Forma farmacéutica del producto.
. Nombre y dirección del titular del registro.
. Fecha de cierre de datos.
. Fecha de elaboración del documento.
. Declaración de confidencialidad y veracidad de la información incluida en el documento.
. Nombre y firma del Responsable de Farmacovigilancia.
II. Tabla de contenidos
III. Parte I: Plan de Farmacovigilancia
III.1 Especificaciones de seguridad:
Debe estar constituido por un resumen de los riesgos de
importancia que se han identificado o que son
potenciales para el producto farmacéutico, y la
información relevante con la que aún no se cuenta.
También debe establecer qué poblaciones se encuentran
en situación potencial de riesgo (cuando es probable
que se utilice el producto), y los asuntos de seguridad
pendientes que requieren mayor investigación para
perfeccionar la comprensión de la relación
beneficio/riesgo durante el período posterior a la
aprobación del producto 7, 8.
El documento de especificaciones de seguridad debe
contener los siguientes elementos:
a. Datos no clínicos 5, 7:
Aspectos de seguridad no clínicos que no han sido
tratados adecuadamente por los datos clínicos, por ejemplo:
. Toxicidad (incluyendo toxicidad a dosis
repetidas, toxicidad reproductiva y del
desarrollo, nefrotoxicidad, hepatotoxicidad,
genotoxicidad, carcinogenicidad, etc.).
. Farmacología general (por ejemplo: cardiovascular,
incluyendo la prolongación del intervalo QT,
sistema nervioso, etc.).
. Interacciones medicamentosas.
. Otros datos o información relacionada con toxicidad.
Si el producto está diseñado para su uso en poblaciones
especiales, se debe tener en cuenta si existen
necesidades específicas de datos no clínicos.
b. Datos clínicos 5:
Limitaciones de la base de datos de seguridad para humanos:
Se deben tomar en cuenta las siguientes limitaciones de
la base de datos de seguridad:
. Tamaño de la población estudiada:
o Dosis, duración.
o Edad, sexo, raza.
o Indicaciones.
. Experiencia en poblaciones especiales.
. Criterios de inclusión/exclusión de los estudios.
. Experiencia postcomercialización a nivel mundial:
o Exposición postcomercialización.
o Riesgos identificados.
o Acciones reguladoras llevadas a cabo.
Poblaciones no estudiadas en la fase de pre-registro:
En las especificaciones se debe discutir las
poblaciones que no se han estudiado o han sido
estudiadas de manera limitada en la fase de
pre-autorización. Debe discutirse explícitamente
las implicaciones que ello tiene con respecto a
la predicción de la seguridad del producto en el
mercado. Las poblaciones que se deben considerar
son, al menos 5, 7, 8:
. Niños.
. Ancianos.
. Mujeres embarazadas o lactantes.
. Pacientes con insuficiencia hepática.
. Pacientes con insuficiencia renal.
. Pacientes con otras co-morbilidades como trastornos
cardíacos o inmunodepresión, incluyendo los
trasplantados.
. Pacientes con enfermedades diferentes o de
gravedad distinta a la estudiada en los
ensayos clínicos.
. Sub-poblaciones portadoras de un polimorfismo
genético conocido y relevante.
. Pacientes de diferentes orígenes raciales
y/o étnicos.
Experiencia postcomercialización 8:
El propósito de este ítem es proveer información
sobre el número de pacientes expuestos
postcomercialización, y mencionar nuevos problemas
de seguridad, particularmente en aquellas
poblaciones previamente no estudiadas. Las
acciones regulatorias de seguridad llevadas a cabo
también deben ser mencionadas.
Eventos adversos/reacciones adversas a medicamentos 7:
. Riesgos identificados que requieren evaluaciones
adicionales: Por lo general, se debe considerar
la necesidad de una evaluación adicional como parte
del Plan de Farmacovigilancia, incluyendo información
más detallada acerca de los EA/RAM más importantes
identificados (graves o serias, frecuentes o los
que podrían tener un impacto en el equilibrio
entre beneficios y riesgos del producto). Esta
información debe incluir la relación causal, la
gravedad, severidad, frecuencia, reversibilidad y
los grupos de riesgo, si están disponibles. Deben
discutirse los factores de riesgo y posibles
mecanismos de los EA/RAM.
. Riesgos potenciales que requieren evaluaciones
adicionales: En esta sección deben describirse los
riesgos potenciales importantes. Debe presentarse
la evidencia que condujo a la conclusión de que
existía un riesgo potencial. Se prevé que para
cualquier riesgo potencial importante debe haber
una evaluación adicional para caracterizar la
asociación.
Tabla Nº 1: Riesgos identificados/potenciales 5, 8.
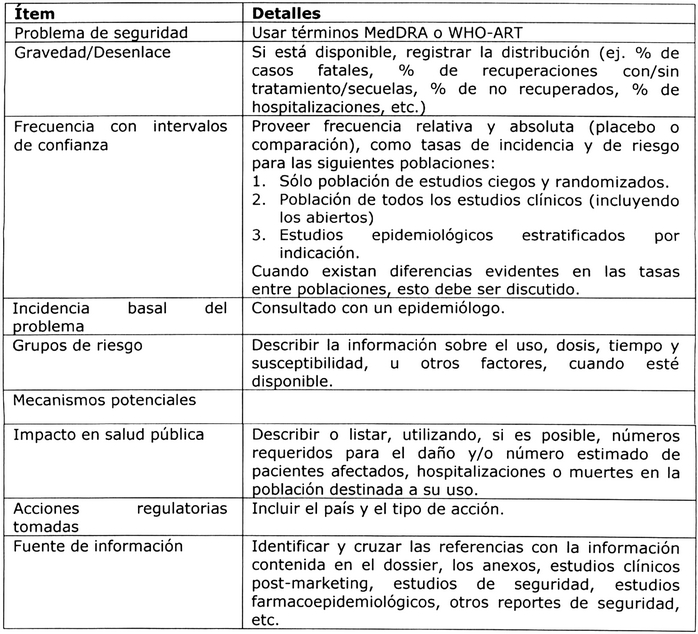
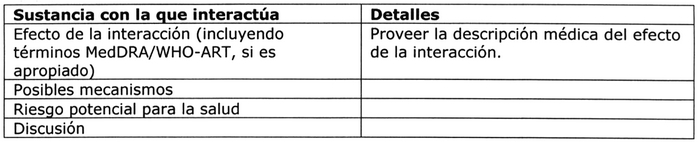
Interacciones identificadas y potenciales, incluyendo
interacciones alimento-fármaco y fármaco-fármaco 7:
Esta sección debe discutir las interacciones
farmacocinéticas y farmacodinámicas identificadas
y potenciales. Para cada una, debe presentarse un
resumen de la evidencia que sustenta la interacción
y sus posibles mecanismos, y deben discutirse
los potenciales riesgos para la salud planteados,
para las diferentes indicaciones y poblaciones.
Tabla Nº 2: Interacciones Medicamentosas 5, 8.
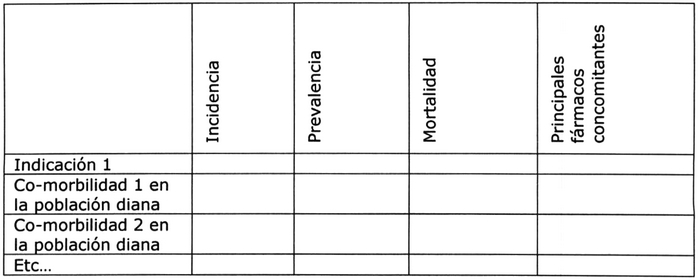
Epidemiología 7:
Esta sección debe discutir la epidemiología de la(s)
indicación(es), incluyendo la incidencia,
prevalencia, mortalidad y co-morbilidad relevantes,
y debe tener en cuenta, siempre que sea posible,
la estratificación por edad, sexo, y el origen
étnico y/o racial. Deben discutirse las diferencias
en la epidemiología de las distintas regiones
(ya que la epidemiología de la(s) indicación(es)
puede(n) variar de una región a otra), si esta
información está disponible.
Además, es útil revisar la incidencia esperable o
basal de ciertos acontecimientos clínicos importantes
que puedan requerir una mayor investigación.
Si está disponible la información sobre factores de
riesgo de un evento adverso, también es de utilidad
incluirla en esta sección.
Tabla Nº 3: Epidemiología: Población diana 5, 8.
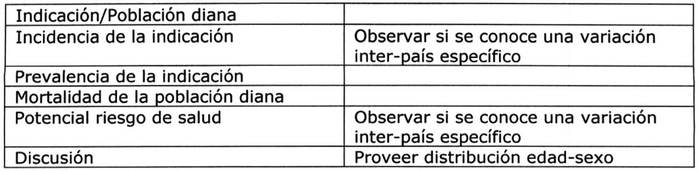
Tabla Nº 4: Epidemiología: Co-morbilidades de la población diana, por indicación 5, 8.
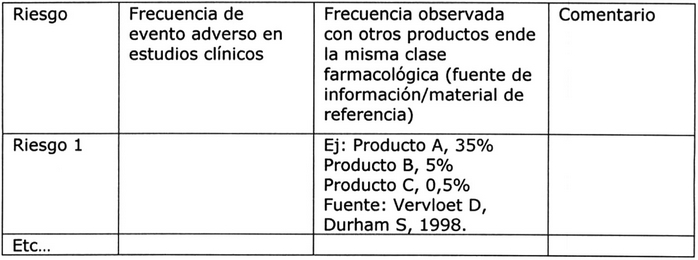
Efectos de la clase farmacológica 7:
Las especificaciones de seguridad deben identificar
los riesgos que son comunes a la clase farmacológica
del producto farmacéutico.
Tabla Nº 5: Efectos de la clase farmacológica 5, 8.
Información adicional 7:
Dependiendo del producto farmacéutico evaluado,
puede ser necesario incorporar algunos de los
siguientes aspectos:
. Potencial de sobredosificación.
. Potencial para la transmisión de agentes infecciosos.
. Potencial para el uso ilegal.
. Potencial para uso fuera de especificaciones de
la monografía o folletos de información.
. Potencial para uso pediátrico fuera de especificaciones.
c. Resumen 7:
Al final de la especificación de seguridad debe incluirse
un resumen de:
. Riesgos importantes identificados.
. Riesgos potenciales importantes.
. Información relevante aún faltante.
Se recomienda resumir los aspectos específicos de seguridad
en un esquema de revisión por tema, incluyendo tanto los
datos clínicos como los no clínicos que tengan
relación con el problema.
III.2. Plan de Farmacovigilancia 7:
El plan de FV debe estar basado en la especificación de seguridad.
Para productos que no presenten problemas especiales bastará con las actividades de rutina de FV.
Para productos que presenten riesgos importantes identificados o potenciales o de los que no se disponga información de seguridad importante, deberán considerarse medidas adicionales destinadas a resolver estos problemas.
Las actividades de FV que se proponen en este plan se incluyen para aportar la información necesaria sobre los riesgos identificados o potenciales señalados en las especificaciones de seguridad, por lo cual deberá actualizarse a medida que se disponga de nueva información importante sobre la seguridad del medicamento y cuando se alcancen hitos preestablecidos.
El plan de FV no incluye acciones con el fin de reducir o prevenir riesgos.
a. Estructura del plan de Farmacovigilancia:
i. Actividades rutinarias de Farmacovigilancia 5, 7:
Debe llevarse a cabo FV de rutina como parte de un
Plan de FV para todos los medicamentos,
independientemente de si es apropiado tomar
medidas adicionales o no. Esta FV de rutina debe
incluir lo siguiente:
. Sistemas y procesos que aseguren que la información
sobre todas las sospechas de reacciones adversas
que se comunican al personal de la empresa son
recopiladas y sistematizadas de una manera accesible.
. Preparación de informes para las autoridades
reguladoras:
o Notificación de RAM en los plazos correspondientes.
o Informes Periódicos de Seguridad.
. Monitorización continuada del perfil de seguridad
de los productos aprobados, incluyendo detección
de señales, evaluación de los problemas,
actualización del etiquetado, y comunicación con
las autoridades reguladoras.
. Otros requerimientos, a solicitud del ISP.
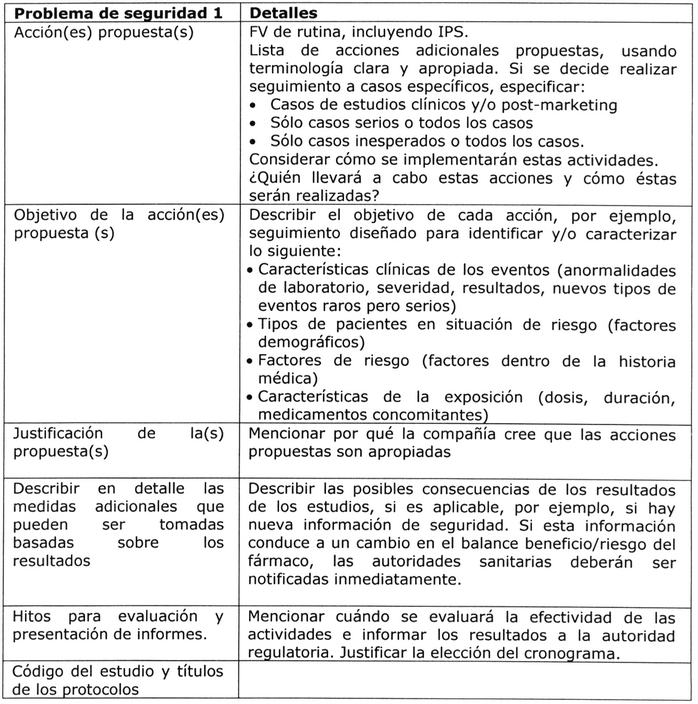
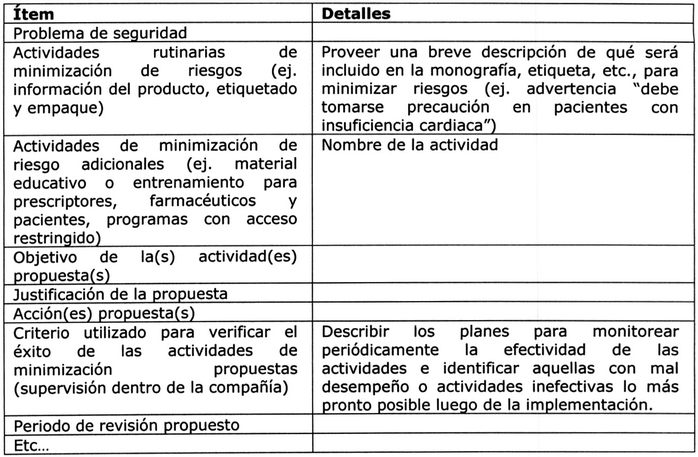
ii. Plan de acción detallado: estudios y otras actividades adicionales:
Debe presentarse y justificarse un plan para cada
problema de seguridad importante de acuerdo con la
siguiente estructura 7:
. Problema de seguridad.
. Acción(es) propuesta(s).
. Objetivo de la(s) acción(es) propuesta(s).
. Justificación de la(s) propuesta(s).
. Supervisión dentro de la compañía.
. Hitos para evaluación y presentación de informes.
El diseño y conducción de estudios farmacoepidemiológicos,
específicamente estudios observacionales
(no intervencional, no-experimental), son
herramientas importantes en farmacovigilancia 7.
Antes de que el estudio observacional forme parte
del plan de farmacovigilancia, el protocolo debe
ser terminado. Es importante contar con la
consultoría de expertos de diferentes disciplinas,
tales como farmacovigilancia, farmacoepidemiología
y bioestadística. Se recomienda que el protocolo
sea discutido con la agencia regulatoria antes de
iniciarlos, al igual si un estudio debe ser
terminado de manera temprana 7.
Los protocolos deben, como mínimo, incluir el
propósito y objetivos del estudio, los métodos que
van a ser utilizados, y el plan para el análisis 7.
Tabla Nº 6: Plan de acción detallado 5, 8.
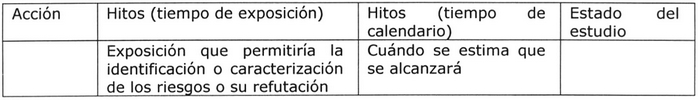
iii. Resumen de las acciones e hitos 5:
En esta sección, el plan de FV para el producto
farmacéutico debe presentarse organizadamente en
términos de las acciones a realizar y sus hitos.
Ello, debido a que una acción propuesta (por ejemplo,
un estudio de cohorte prospectivo de seguridad)
podría abordar más de uno de los problemas
identificados.
Se recomienda que en el plan de FV se incluyan
los hitos para la realización de estudios y
evaluaciones, y para la presentación de los
resultados de seguridad. En la determinación de
estos hitos se debe considerar:
. Tiempo de exposición que permitirá la identificación
o caracterización de los riesgos o su refutación.
. Tiempo en el cual se estima que se alcanzará
y estarán disponibles los resultados de los
estudios de seguridad en curso o en proyecto.
Los hitos pueden fijarse conforme a los tiempos
de los requerimientos reguladores ordinarios
(ej. IPS, renovaciones, etc.).
Tabla Nº 7: Resumen de las acciones e hitos 5, 8.
iv. Evaluación de la necesidad de elaborar un plan de
minimización de riesgos 5, 8.
Esta sección debe tener una discusión sobre la necesidad
o no de un plan de minimización de riesgos adicional
al plan de farmacovigilancia. Debe realizarse una
revisión de los problemas reales o potenciales,
relacionados a errores de medicación, sobredosis, uso
pediátrico, uso off-label, transmisión de agentes
infecciosos, abuso, entre otros.
Si es necesario incluir medidas adicionales para que
el perfil del producto farmacéutico mantenga su
relación beneficio/riesgo positiva, será necesario
planificar una estrategia de minimización de riesgos.
Tabla Nº 8: Evaluación de necesidad de medidas de minimización 5.
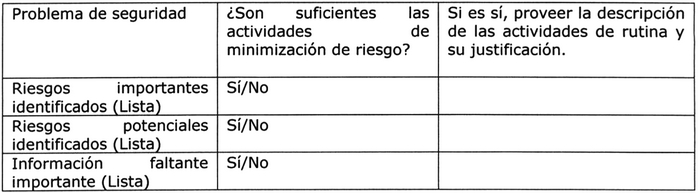
IV. Parte II: Plan de minimización de riesgos
IV.1 Plan de minimización de riesgos
Para cada problema de seguridad identificado en el resumen de las especificaciones de seguridad, debe entregarse la siguiente información 5, 7:
. Problema de seguridad.
. Actividades rutinarias de minimización de riesgos.
. Actividades adicionales de minimización de riesgos.
. Objetivo de la(s) acción(es) propuesta(s).
. Justificación de la(s) propuesta(s).
. Acción(es) propuesta(s).
. Criterio utilizado para verificar el éxito de las actividades de minimización propuestas (supervisión dentro de la compañía)
. Período de revisión propuesto.
Es importante considerar que un problema puede requerir más de una medida, como también una misma medida puede abordar más de un problema de seguridad 7.
Tabla Nº 9: Plan de minimización de riesgos 5, 8.
IV.2 Evaluación de la efectividad de las medidas e intervenciones 5:
El éxito de las medidas de minimización de riesgos requiere ser evaluado a través de todo el ciclo de vida del producto farmacéutico para asegurar que la carga de las RAM sea minimizada y, por lo tanto, el perfil de seguridad sea optimizado. Cuando el plan de manejo de riesgos es actualizado, el plan de minimización de riesgos debe incluir una evaluación del impacto de las actividades de minimización rutinarias y adicionales. Como parte de esta evaluación crítica, el TRS debe hacer observaciones sobre el éxito o pobre desempeño de las actividades de minimización. Si una estrategia particular de minimización de riesgos prueba ser inefectiva, o estar causando una carga excesiva o indebida en los pacientes o el sistema de salud, entonces se requiere poner en su lugar actividades alternativas. Los TRS siempre deben comentar si son necesarias actividades de minimización de riesgos adicionales o diferentes para cada problema de seguridad planteado. En ciertos casos, puede juzgarse que la minimización de riesgos no puede controlar los riesgos lo suficiente para asegurar un balance beneficio/riesgo positivo y que el producto farmacéutico necesita ser retirado del mercado o restringido a aquellos pacientes en los cuales los beneficios superan los riesgos.
Para esta evaluación serán esenciales fuentes de información, tales como:
. Bases de datos de prescripción/dispensación.
. Estudios "ad hoc": Encuestas.
. Bases de datos clínicas.
. Registros permanentes.
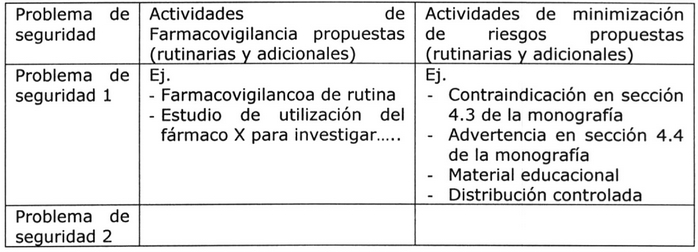
V. Resumen del Plan de Manejo de Riesgos 5:
En este ítem se debe proporcionar una tabla resumen con todas las acciones de farmacovigilancia y de minimización de riesgos, tanto de rutina como adicionales.
Tabla Nº 10: Resumen del Plan de Manejo de Riesgos 5.
VI. Referencias:
Se deben incluir las referencias bibliográficas utilizadas en la elaboración del documento.
VII. Anexos:
Se debe incluir toda la información que se estime pertinente anexar al documento.
ENVÍO DEL PLAN DE MANEJO DE RIESGOS AL SUBDEPARTAMENTO DE FARMACOVIGILANCIA
El titular del registro puede preparar un único Plan de Manejo de Riesgos para todos los productos farmacéuticos que contengan el mismo principio activo. Estos deben ser presentados cuando la autoridad sanitaria lo solicite, según resolución, en formato digital a través del mail cenimef@ispch.cl, o bien por Oficina de Partes. El documento debe presentarse íntegramente en idioma español.
Las situaciones en las que podrá ser necesaria su presentación son:
. Moléculas introducidas por primera vez en el mercado;
. Producto de origen biológico y sus similares (biosimilares);
. Medicamento genérico si es que el de referencia está sometido a actividades de minimización de riesgos;
. Otros casos calificados, según se establece en el artículo 218 del decreto supremo Nº 3/2010.
Referencias
1. Minsal. Decreto supremo Nº 3/2010. Reglamento del Sistema Nacional de Control de los Productos Farmacéuticos de Uso Humano.
2. Minsal. Norma General Técnica Nº 140 sobre Sistema Nacional de Farmacovigilancia de Productos Farmacéuticos de Uso Humano. 2012.
3. Instituto de Salud Pública. Resolución exenta Nº 441. Establece y actualiza el proceso de notificación de eventos adversos ocurridos en ensayos clínicos que se desarrollan en Chile. 2012.
4. OPS. Documento Técnico Nº 5 de la Red Panamericana de Armonización de la Reglamentación Farmacéutica, "Buenas Prácticas de Farmacovigilancia para las Américas". 2010.
5. Ema. Guideline on good pharmacovigilance practices (GVP). Module V Risk management systems. 2012.
6. ANMAT. Guía de Buenas Prácticas de Farmacovigilancia. 2009.
7. ICH. Pharmacovigilance Planning E2E. 2004.
8. Anvisa. Regulatory Guide. Pharmacovigilance Plan and Risk Minimization Plan PVP/RMP. 2009.
Anótese, comuníquese y publíquese la presente resolución en el Diario Oficial, sin perjuicio de su inclusión en la página web institucional por parte de la Unidad de Comunicaciones e Imagen Institucional.- Stephan Jarpa Cuadra, Director (S).
documento impreso desde www.bcn.cl/leychile
el 18 del
05 de 2025
a las 18 horas con
2
minutos.