DETERMINA LOS DIAGNÓSTICOS Y TRATAMIENTOS DE ALTO COSTO CON SISTEMA DE PROTECCIÓN FINANCIERA DE LA LEY Nº 20.850
Núm. 2.- Santiago, 18 de enero de 2019.
Vistos:
El DFL Nº 1, de 2005, del Ministerio de Salud; la ley Nº 20.850; el decreto supremo Nº 59, de 2015, de los Ministerios de Salud y de Hacienda, que aprueba el Reglamento que establece el procedimiento para fijar el umbral nacional de costo anual para determinar si un diagnóstico o un tratamiento son de alto costo; el decreto supremo Nº 54, de 2015, del Ministerio de Salud, que establece normas para el otorgamiento y cobertura financiera de los diagnósticos y tratamientos incorporados al sistema establecido en la ley Nº 20.850; el decreto supremo Nº 86, de 2018, de los Ministerios de Salud y de Hacienda, que fija el umbral nacional de costo anual para determinar si un diagnóstico o un tratamiento son de alto costo; decreto supremo Nº 13, de 2017, que aprueba el reglamento que establece el proceso destinado a determinar los diagnósticos y tratamientos de alto costo con sistema de protección financiera, según lo establecido en los artículos 7º y 8º de la ley Nº 20.850; y la resolución Nº 1.600, de 2008, de la Contraloría General de la República, y
Considerando:
1º Que, con fecha 6 de junio de 2015, se publicó la ley Nº 20.850, que crea un Sistema de Protección Financiera para Diagnósticos y Tratamientos de Alto Costo y rinde homenaje póstumo a don Luis Ricarte Soto Gallegos.
2º Que, en virtud de lo dispuesto en el artículo 5º de ese cuerpo legal, los diagnósticos y tratamientos asociados a condiciones específicas de salud que se incorporan al Sistema de Protección Financiera, deben ser determinados a través de un decreto supremo del Ministerio de Salud, suscrito también por el Ministro de Hacienda.
3º Que, conforme al Reglamento sobre Umbral Nacional de Costo Anual, aprobado mediante decreto supremo Nº 59, de los Ministerios de Salud y Hacienda y al artículo 6º de la ley, se dictó el decreto supremo Nº 86, de 2018, de los Ministerios de Salud y Hacienda, determinando el Umbral en $3.011.381.- (tres millones once mil trescientos ochenta y un pesos).
4º Que, a través del decreto supremo Nº 13, de 2017, del Ministerio de Salud, se aprobó el reglamento que establece el proceso destinado a determinar los diagnósticos y tratamientos de alto costo con sistema de protección financiera, según lo establecido en los artículos 7º y 8º de la ley Nº 20.850, en adelante el Reglamento.
5º Que, a través del decreto exento Nº 345, de 2017, del Ministerio de Salud, se aprueba Norma Técnica Nº 192, sobre el proceso de evaluación científica de la evidencia establecido en el artículo 7º de la ley Nº 20.850.
6º Que, en conformidad con lo dispuesto en el Título II del Reglamento, fueron recibidas solicitudes de evaluación de tecnologías sanitarias, las que se encuentran disponibles en el sitio electrónico http://web.minsal.cl/ley-ricarte-solicitudes-ciudadanas/.
7º Que, mediante la resolución exenta Nº 1.070, de 2018, del Ministerio de Salud, y, de acuerdo a lo establecido en el artículo 26º del reglamento citado en el considerando 4º, se individualizó a todos los participantes en la etapa de evaluación científica de la evidencia.
8º Que, considerando lo dispuesto en los artículos 6º y 9º del Reglamento, a través de las resoluciones exentas Nº 840 y 1.036, de 2018, del Ministerio de Salud, se dio inicio a la evaluación científica de la evidencia para los tratamientos en ellas señalados, procedimiento que fue desarrollado en conformidad con lo prescrito en el Título III del Reglamento.
9º Que, para la realización de los informes de evaluación científica de la evidencia, fue solicitada información a posibles proveedores de las tecnologías objeto de evaluación, de acuerdo a lo dispuesto en el artículo 17 del Reglamento.
10º Que, los informes favorables de evaluación científica de la evidencia fueron publicados en http://web.minsal.cl/ley-ricarte-soto-proceso-de-evaluacion/.
11º Que, sobre la base de dichos informes, se dio inicio a la etapa de Recomendación Priorizada de la ley Nº 20.850, la que fue desarrollada en conformidad con lo dispuesto en el Título IV, del Reglamento.
12º Que, para el desarrollo de esta etapa, a través de la resolución exenta Nº 1.171, de 2017, del Ministerio de Salud, se creó el Comité de Evaluación de Postulantes para la integración de la Comisión de Recomendación Priorizada establecida en el artículo 8º de la ley Nº 20.850, cuyos miembros fueron designados a través de la resolución exenta Nº 606, de 2018, del Ministerio de Salud.
13º Que, por medio de la resolución exenta Nº 1.000, de 2017, del Ministerio de Salud, se dio inicio al proceso de selección de representantes de las agrupaciones o asociaciones de pacientes en la Comisión de Recomendación Priorizada.
14º Que, por resolución exenta Nº 1.254, de 2017, del Ministerio de Salud, se designó a los integrantes para conformar la Comisión de Recomendación Priorizada, conformación que fue modificada posteriormente a través de las resoluciones exentas Nº 141 y 920, de 2018, del Ministerio de Salud.
15º Que, a través de la resolución exenta Nº 1.077, de 2018, del Ministerio de Salud, se aprobó la metodología de priorización a ser utilizada por la Comisión de Recomendación Priorizada establecida en el artículo 8º de la ley Nº 20.850.
16º Que, en virtud de la aplicación de dicha metodología, con fecha 23 de octubre de 2018 fue publicada en el sitio electrónico http://web.minsal.cl/comision-recomendacion-priorizada-ley-ricarte-soto/, el acta final que contiene las conclusiones del proceso deliberativo realizado por la Comisión.
17º Que, en conformidad con lo dispuesto en el artículo 8º de la ley y el Párrafo 6º del Título IV del ya citado reglamento, fueron recibidos recursos de impugnación en contra de la referida acta final.
18º Que, para la resolución de los recursos señalados, la Comisión sesionó con fecha 31 de octubre, 5, 6, 9 y 12 de noviembre, resolviendo la totalidad de ellos, según lo dispuesto en el artículo 63 del reglamento citado en el considerando 4º.
19º Que, en virtud de lo anterior, con fecha 12 de noviembre fue levantada el Acta final Resolutiva de los Recursos de Impugnación y Priorización Final de la Comisión de Recomendación Priorizada, la que se encuentra publicada en el sitio electrónico del Ministerio de Salud, junto a cada acta que resolvió los indicados recursos.
20º Que, el inciso tercero del artículo 9º de la ley Nº 20.850, dispone que "el conjunto de Tratamientos de Alto Costo que cubrirá el Sistema de Protección Financiera deberá tener un costo anual esperado, para el período de vigencia del correspondiente decreto, igual o inferior al ochenta por ciento del valor esperado al 1 de enero del año siguiente a su dictación, de los recursos totales con que contará el Fondo en dicho año". De este modo, para este cuarto decreto, el costo anual esperado de los diagnósticos y tratamientos de alto costo a incluir no puede ser superior al 80% de los recursos totales con que contará el fondo para el período comprendido entre los años 2019 a 2022.
21º Que, para estos efectos conforme a lo dispuesto en el inciso final del artículo 6º, del reglamento de la especie, el Ministerio de Hacienda, a través de Ordinario Nº 2171, de 2018, del Director de Presupuestos, informó la disponibilidad presupuestaria para el periodo de vigencia del presente decreto.
22º Que, sin perjuicio de lo anterior, para los efectos de la dictación del presente decreto y de lo dispuesto en el artículo 9º de la ley, a través de correo electrónico de fecha 11 de enero del presente año, el Director de Presupuestos del Ministerio de Hacienda informó una holgura adicional del Fondo de, aproximadamente, diez mil millones de pesos.
23º Que, cada uno de los informes de evaluación publicados en el sitio electrónico del Ministerio de Salud, contiene un apartado respecto a la consideración de la existencia de redes asistenciales disponibles de forma inmediata que cumplen con los requisitos establecidos en el reglamento aprobado mediante decreto supremo Nº 54, de 2015, del Ministerio de Salud, para los nuevos diagnósticos y tratamientos de alto costo que ingresan al sistema de protección financiera el 1 de julio de 2019, apartado elaborado sobre la base de lo informado por la Subsecretaría de Redes Asistenciales, dándose cumplimiento de esta forma a lo establecido en el inciso séptimo del artículo 7º de la ley Nº 20.850.
24º Que, finalmente, se consideró el costo de los tratamientos y la disponibilidad de los recursos en el Fondo, de manera de dar cumplimiento a las disposiciones contenidas en el artículo 9º de la ley Nº 20.850.
25º Que, sin perjuicio de la priorización de diagnósticos y tratamientos de alto costo realizada por la Comisión destinada para dicha función, los Ministerios de Salud y de Hacienda, en virtud de lo establecido en el artículo 9º de la ley Nº 20.850, han decidido ampliar la cobertura de profilaxis con Palivizumab para lactantes menores de 1 año con cardiopatías congénitas hemodinámicamente significativas, en razón que la cobertura ya existente para pacientes prematuros con y sin displasia broncopulmonar tiene un efecto de impacto absoluto similar al de este grupo, aumentando así la equidad asociada a la cobertura de esta intervención.
26º Que, además, es necesario ampliar la cobertura para personas con Esclerosis Múltiple Recurrente Remitente con falla a tratamiento con inmunomoduladores con los fármacos Alemtuzumab, Cladribina y Ocrelizumab dado que estos tratamientos presentan eficacia comprobada para este grupo de beneficiarios y, además, un impacto presupuestario por paciente menor a aquellos fármacos ya cubiertos por el Fondo, por lo que su incorporación al sistema de protección financiera significaría no solo una ampliación del arsenal terapéutico, sino también un potencial menor impacto presupuestario para el Fondo.
27º Que, en ese sentido, se hace necesario ampliar la cobertura para personas con Artritis Reumatoide activa refractaria a tratamiento habitual con los fármacos Tocilizumab, Tofacitinib y Golimumab, dado que estos tratamientos presentan eficacia comprobada para este grupo de beneficiarios y, además, un impacto presupuestario por paciente menor a aquellos fármacos ya cubiertos por el fondo, por lo que su incorporación al Sistema de Protección Financiera significaría no solo una ampliación del arsenal terapéutico, sino también un potencial menor impacto presupuestario para el Fondo.
28º Que, por su parte, se hace necesario incorporar el tratamiento con Ocrelizumab para personas con Esclerosis Múltiple Primaria Progresiva, en consideración de que este tratamiento presenta eficacia comprobada para este grupo de personas, no habiendo en la actualidad otra alternativa terapéutica efectiva, considerando además que este tratamiento será incorporado como alternativa terapéutica para personas con Esclerosis Múltiple Recurrente Remitente con falla a tratamiento con inmunomoduladores, por lo que su inclusión en el grupo de personas con Esclerosis Múltiple Primaria Progresiva, aumenta la equidad asociada a la cobertura de esta intervención.
29º Que, finalmente, se hace necesario incorporar el tratamiento con inmunoglobulina G subcutánea para personas con Inmunodeficiencia Primaria, en consideración de que esta alternativa de tratamiento es de utilidad en personas que no pueden recibir inmunoglobulina G endovenosa debido a la falta de accesos vasculares o a reacciones adversas a ésta, mejorando de esta forma la integralidad de la atención para este problema de salud.
30º Que, de este modo, la elección de los nuevos tratamientos a incluir en este decreto se ajusta a las disposiciones de la ley Nº 20.850, los cuales cumplieron estrictamente con los criterios establecidos en el proceso de evaluación y priorización establecido en el reglamento aprobado por decreto Nº 13, de 2017, del Ministerio de Salud. Asimismo, su impacto fiscal en la red pública de salud resulta acorde con lo asignado para dichos fines en la Ley de Presupuestos del Sector Público vigente y son coherentes con la cobertura poblacional y de prestaciones en el sistema público de salud.
31º Que, los demás diagnósticos y tratamientos de alto costo incorporados al presente decreto, formaron parte del decreto Nº 87, de 2015, del decreto Nº 50, de 2016, y del decreto Nº 47, de 2017, todos del Ministerio de Salud, por lo que, de acuerdo a lo dispuesto en el inciso segundo del artículo tercero transitorio del reglamento indicado en el considerando 4º, los diagnósticos y tratamientos aprobados en dichos decretos se entenderán prorrogados durante el período de vigencia del presente decreto.
32º Que, algunos de los precios informados a este Ministerio en virtud de lo dispuesto en el artículo 17 del Reglamento, han sido actualizados, pues los proveedores hicieron ofertas que, en razón de la oportunidad de su presentación, no fueron los considerados en los informes de evaluación de los tratamientos, pero que generan una situación provechosa para el Fondo, pues son precios más económicos.
33º Que, en conformidad al inciso segundo del artículo 9º de la ley Nº 20.850, la Dirección de Presupuesto emitió el Informe de Sustentabilidad Financiera, que se acompaña al presente decreto, el cual demuestra que el costo anual de todos los tratamientos señalados en este decreto no superan anualmente el 80% de los recursos del Fondo, ajustándose a lo dispuesto en el artículo en comento.
34º Que, mediante resoluciones exentas Nº 735, de 2015, Nº 1.447, de 2016, y Nº 1.664, de 2017, todas del Ministerio de Salud, se aprobaron protocolos para tratamientos asociados a enfermedades o condiciones de salud específicas, que se incorporan en este decreto y que ya fueron incluidas en la cobertura financiera durante el año 2016, 2017, 2018 y hasta el 30 de junio de 2019, respectivamente.
35º Que, además, mediante resolución exenta Nº 38, de 17 de enero de 2019, del Ministerio de Salud, se aprobaron los protocolos para cada uno de los tratamientos asociados a enfermedades o condiciones de salud específicas que se incorporarán a la cobertura desde el 1 de julio del año 2019.
36º Que, conforme a lo dispuesto en el artículo primero transitorio de la ley Nº 20.850, el decreto Nº 47, de 2017, tiene vigencia hasta el 30 de junio de 2019, por lo que los diagnósticos y tratamientos ahí garantizados, dentro de los cuales se incluyen los incorporados por el decreto supremo Nº 87, de 2015, y Nº 50, de 2016, ambos del Ministerio de Salud, son incorporados al presente decreto en las condiciones en él especificadas.
37º Que, habiéndose cumplido los requisitos y procedimientos establecidos en la ley, dicto el siguiente:
Decreto:
1º Determínanse como diagnósticos y tratamientos de alto costo para condiciones específicas de salud con sistema de protección financiera de la ley Nº 20.850 los siguientes:
1. Decreto 36,
SALUD
N° 1
D.O. 13.01.2025DIAGNÓSTICO Y TRATAMIENTO BASADO EN LARONIDASA PARA PERSONAS CON ENFERMEDAD DE MUCOPOLISACARIDOSIS TIPO I
SALUD
N° 1
D.O. 13.01.2025DIAGNÓSTICO Y TRATAMIENTO BASADO EN LARONIDASA PARA PERSONAS CON ENFERMEDAD DE MUCOPOLISACARIDOSIS TIPO I
La Mucopolisacaridosis tipo I es una enfermedad del grupo de los errores innatos del metabolismo lisosomal, de carácter autosómica recesiva. Es causada por una acumulación progresiva de sustratos complejos de glucosaminoglucanos, dermatán y heparán sulfato, debido a la deficiencia de la enzima alfa-L-iduronidasa. Este depósito lisosomal se produce en una amplia variedad de órganos, lo que conlleva a una disfunción multiorgánica debilitante y fatal, con presentación clínica variable.
a) Población beneficiaria
Personas con sospecha o diagnóstico de Mucopolisacaridosis tipo I que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1 Confirmación diagnós tica
Examen de medición de la actividad enzimática en fibroblastos o leucocitos, o examen genético molecular según indicación para el diagnóstico.
b.2 Tratamiento
Medicamento Laronidasa para terapia de reemplazo enzimático.
c) Garantías de oportunidad
c.1. Confirmación Diagnóstica
El procesamiento de la muestra y resultado de examen de medición de la actividad enzimática en fibroblastos o leucocitos se realizará dentro de 20 días desde la recepción del formulario de sospecha fundada y la muestra de leucocitos para determinación enzimática por el prestador aprobado para la etapa de confirmación.
El examen genético molecular se realizará, en caso de existir dos exámenes de determinac ión de actividad enzimática con resultado indeterminado, dentro de 90 días desde la recepción de la muestra por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Laronidasa se realizará con confirmación diagnóstica y cumplimiento de los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento con Laronidasa, previo a ser beneficiario de esta ley, y cumpliendo con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
2. DIAGNÓSTICO Y TRATAMIENTO BASADO EN IDURSULFASA PARA PERSONAS CON ENFERMEDAD DE MUCOPOLISACARIDOSIS TIPO II
La Mucopolisacaridosis tipo II o Síndrome de Hunter es un trastorno hereditario que presenta un patrón de herencia ligada al cromosoma X y es causada por el déficit de la enzima iduronatosulfatasa que participa en la degradación de dermatán sulfato y heparansulfato.
En la Mucopolisacaridosis tipo II se acumulan cantidades perjudiciales de glucosaminoglucanos, dermatán y heparán sulfato en la matriz extracelular del tejido conectivo. Esta acumulación es progresiva, por lo que con el tiempo los síntomas se evidencian con mayor severidad.
a) Población beneficiaria
Personas con sospecha o diagnóstico de Mucopolisacaridosis tipo II que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de medición de la actividad enzimática en fibroblastos o leucocitos, o examen genético molecular según indicación para el diagnóstico.
b.2. Tratamiento
Medicamento Idursulfasa para terapia de reemplazo enzimático.
c) Garantías de oportunidad
c.1. Confirmación Diagnóstica
El procesamiento de la muestra y resultado de examen de medición de la actividad enzimática en fibroblastos o leucocitos se realizará dentro de 20 días desde la recepción del formulario de sospecha fundada y la muestra de leucocitos para determinación enzimática por el prestador aprobado en la etapa de confirmación.
El examen genético molecular se realizará, en caso de existir dos exámenes de determinación de actividad enzimática con resultado indeterminado, dentro de 90 días desde la recepción de la muestra por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Idursulfasa se realizará con confirmación diagnóstica y cumplimiento de los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento con Idursulfasa, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestacione s de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
3. DIAGNÓSTICO Y TRATAMIENTO BASADO EN GALSULFASA PARA PERSONAS CON ENFERMEDAD DE MUCOPOLISACARIDOSIS TIPO VI
La Mucopolisacaridosis tipo VI es una enfermedad autosómica recesiva causada por la deficiencia de la enzima lisosomal Arilsulfatasa B o N -acetilgalactosamina-4-sulfatasa. Esto da como resultado la acumulación patológica de dermatán sulfato a nivel celular en distintos tejidos.
a) Población beneficiaria
Personas con sospecha o diagnóstico de Mucopolisacaridosis tipo VI que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de medición de la actividad enzimática en fibroblastos o leucocitos, o examen genético molecular según indicación para el diagnóstico.
b.2. Tratamiento
Medicamento Galsulfasa para terapia de reemplazo enzimático.
c) Garantías de oportunidad
c.1. Confirmación Diagnóstica
El procesamiento de la muestra y resultado de examen de medición de la actividad enzimática en fibroblastos o leucocitos se realizará dentro de 20 días desde la recepción del formulario de sospecha fundada y la muestra de leucocitos para determinación enzimática por el prestador aprobado en la etapa de confirmación.
El examen genético molecular se realizará, en caso de existir dos exámenes de determinación de actividad enzimática con resultado indeterminado, dentro de 90 días desde la recepción de la muestra por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Galsulfasa se realizará con confirmación diagnóstica y cumplimiento de los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, dentro de 60 días desde la solicitud del tratamiento. Estando en tratamiento con Galsulfasa, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
4. DIAGNÓSTICO Y TRATAMIENTO BASADO EN NITISINONA PARA PERSONAS CON TIROSINEMIA TIPO I
La tirosinemia tipo I es una enfermedad metabólica que se produce por el déficit de las enzimas fumarilacetoacetasa hidrolasa, produciendo la acumulación de fumarilacetoacetato y maleilacetoacetato que serían agentes productores del daño hepatorrenal.
a) Población beneficiaria
Personas con sospecha o diagnóstico de tirosinemia tipo I que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de determinación de niveles elevados de succinilacetona en plasma u orina.
b.2. Tratamiento
Medicamento Nitisinona para terapia de reemplazo enzimático
c) Garantías de oportunidad
c.1. Confirmación Diagnóstica
El procesamiento de la muestra y resultado de examen de medición por determinación de niveles de succinilacetona en plasma u orina se realizará dentro de 4 días hábiles desde la recepción del formulario de sospecha fundada y la muestra de sangre u orina, por el prestador aprobado en la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Nitisinona se realizará con sospecha clínica fundada y laboratorio compatible (tirosina elevada por espectrometría de masa en tándem de papel filtro o succinilacetona elevada en plasma u orina por cromatografía de gases, espectrometría de masa (CG/MS)), dentro de 48 horas desde la recepción del formulario de sospecha fundada y de la muestra de sangre u orina por el prestador aprobado en la etapa de tratamiento. Con diagnóstico confirmado de tirosinemia tipo 1 tendrá acceso a continuar el tratamiento con Nitisinona.
Estando en tratamiento con Nitisinona, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 48 horas, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
5. TRATAMIENTO DE SEGUNDA LÍNEA BASADO EN FINGOLIMOD O NATALIZUMAB O ALEMTUZUMAB O CLADRIBINA U OCRELIZUMAB U OFATUMUMAB PARA PERSONAS CON ESCLEROSIS MÚLTIPLE RECURRENTE REMITENTE CON FALLA A TRATAMIENTO CON INMUNOMODULADORES Y TRATAMIENTO CON OCRELIZUMAB PARA PERSONAS CON ESCLEROSIS MÚLTIPLE PRIMARIA PROGRESIVA
La Esclerosis Múltiple, corresponde a una enfermedad desmielinizante del sistema nervioso central, que se manifiesta con variada sintomatología deficitaria según el territorio anatómico afectado.
a) Población beneficiaria
Personas con diagnóstico de esclerosis múltiple recurrente remitente con falla a tratamiento con inmunomoduladores, o con diagnóstico de esclerosis múltiple primaria progresiva, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento Fingolimod o Natalizumab o Alemtuzumab o Cladribina u Ocrelizumab u Ofatumumab, para tratamiento farmacológico de esclerosis múltiple recurrente remitente con falla a tratamiento con inmunomoduladores.
Medicamento Ocrelizumab, para tratamiento farmacológico de esclerosis múltiple primaria progresiva.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas que tengan confirmación del diagnóstico de esclerosis múltiple recurrente remitente con falla a tratamiento con inmunomoduladores, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Fingolimod o Natalizumab o Alemtuzumab o Cladribina u Ocrelizumab u Ofatumumab, según la indicación del médico tratante registrada al momento de generar la solicitud, dentro de 60 días desde la solicitud del tratamiento.
Las personas que tengan confirmación del diagnóstico de esclerosis múltiple primaria progresiva que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Ocrelizumab dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
6. DIAGNÓSTICO Y TRATAMIENTO BASADO EN TALIGLUCERASA O IMIGLUCERASA PARA PERSONAS CON ENFERMEDAD DE GAUCHER
La enfermedad de Gaucher es una enfermedad que se produce por el déficit de la enzima lisosomal glucocerebrosidasa que se transmite de manera autosómica recesiva. Se caracteriza por el compromiso visceral, hematológico y óseo.
a) Población beneficiaria
Personas con sospecha o diagnóstico de enfermedad de Gaucher, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de medición de la actividad enzimática en leucocitos o examen genético molecular según indicación.
b.2. Tratamiento
Medicamento Taliglucerasa o Imiglucerasa para terapia de reemplazo enzimático.
c) Garantías de oportunidad
c.1. Confirmación Diagnóstica
El procesamiento de la muestra y resultado de examen de medición de la actividad enzimática en leucocitos se realizará dentro de 21 días desde la recepción del formulario de sospecha fundada y la muestra de leucocitos para determinación enzimática, por el prestador aprobado en la etapa de confirmación.
El examen genético molecular se realizará, en caso de existir dos exámenes de determinación de actividad enzimática con resultado indeterminado, dentro de 90 días desde la recepción de la muestra por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Taliglucerasa o Imiglucerasa, se realizará con confirmación diagnóstica y cumplimiento de los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento con Taliglucerasa o Imiglucerasa, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
7. DIAGNÓSTICO Y TRATAMIENTO BASADO EN AGALSIDASA PARA PERSONAS CON ENFERMEDAD DE FABRY
La enfermedad de Fabry es una enfermedad multisistémica, crónica, progresiva, de carácter hereditario y ligada al cromosoma X. El déficit enzimático es consecuencia de una mutación en el gen de la a-galactosidasa A, determinando el depósito de glucoesfingolípidos neutros, que se acumulan en los lisosomas de diversos tejidos. El carácter progresivo de su evolución natural ocasiona una serie de complicaciones graves principalmente renales y cardiacas que reducen la expectativa y calidad de vida.
a) Población beneficiaria
Personas con sospecha o diagnóstico de enfermedad de Fabry, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de medición de la actividad enzimática en leucocitos o examen genético molecular según indicación para el diagnóstico.
b.2. Tratamiento
Medicamento Agalsidasa alfa y beta para terapia de reemplazo enzimático.
c) Garantías de oportunidad
c.1. Confirmación diagnóstica
En personas de sexo masculino, el procesamiento de la muestra y resultado del examen de medición de la actividad enzimática en leucocitos se realizará dentro de 30 días desde la recepción del formulario de sospecha fundada y la muestra de leucocitos para determinación enzimática por el prestador aprobado en la etapa de confirmación. En caso de existir dos exámenes de determinación de actividad enzimática con resultado indeterminado se realizará el examen genético molecular, dentro de 90 días desde la recepción de la muestra por el prestador aprobado para la etapa de confirmación.
En personas de sexo femenino, se realizará el examen genético molecular dentro de 30 días desde la recepción del formulario de sospecha fundada por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Agalsidasa alfa o beta, se realizará con confirmación diagnóstica y cumplimiento de los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento con Agalsidasa alfa o beta, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
8. DIAGNÓSTICO Y TRATAMIENTO BASADO EN ILOPROST INHALATORIO O AMBRISENTÁN O BOSENTÁN PARA PERSONAS CON HIPERTENSIÓN ARTERIAL PULMONAR GRUPO I
Se define hipertensión pulmonar arterial (HAP) grupo I del punto de vista hemodinámico invasivo, como el aumento de la presión media de la arteria pulmonar ≥ 25 mmHg (PAPm ≥ 25 mmHg) con capilar pulmonar ≤ 15mmHg. Es una enfermedad crónica y progresiva, de baja prevalencia, pero alto impacto por su curso grave y potencialmente letal.
a) Población beneficiaria
Personas con sospecha o diagnóstico de hipertensión arterial pulmonar del grupo I, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen de cateterismo cardiaco según indicación para el diagnóstico.
b.2. Tratamiento
Medicamento Iloprost inhalatorio o Ambrisentán o Bosentán.
c) Garantías de oportunidad
c.1. Confirmación diagnóstica
Con sospecha clínica fundada, e indicación médica, se realizará examen de cateterismo cardiaco dentro de 40 días hábiles desde la recepción del formulario de sospecha fundada por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
En personas mayores de 18 años que tengan confirmación del diagnóstico de hipertensión arterial pulmonar del grupo I y falla a tratamiento de primera línea, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Ambrisentan o Bosentan, o en terapia combinada con Iloprost inhalatorio, según la indicación del médico tratante registrada al momento de generar la solicitud, dentro de 15 días desde la solicitud del tratamiento. En el caso de estar hospitalizado en UCI el inicio del tratamiento será en 72 horas, desde la solicitud del tratamiento.
En personas menores de 18 años que tengan confirmación del diagnóstico de hipertensión arterial pulmonar del grupo I y falla a tratamiento de primera línea que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Bosentan, según la indicación del médico tratante registrada al momento de generar la solicitud, dentro de 15 días desde la solicitud del tratamiento.
En el caso de estar hospitalizado en UCI el inicio del tratamiento será en 72 horas, desde la solicitud del tratamiento. Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 15 días desde la solicitud del tratamiento y en caso de estar hospitalizado en UCI dentro de 72 horas, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
9. TRATAMIENTO BASADO EN TRASTUZUMAB PARA PERSONAS CON CÁNCER DE MAMA QUE SOBREEXPRESAN EL GEN HER2 (HER2+)
El cáncer de mama es una enfermedad en su mayoría hormono dependiente debido al crecimiento anormal y desordenado de células del epitelio de los conductos o lobulillos mamarios y que tiene la capacidad de diseminarse. Aproximadamente el 25% de los cánceres de mama son tipo HER2 positivo, el cual tiende a ser más agresivo, de peor pronóstico y con mayores tasas de recaída.
a) Población beneficiaria
Personas con diagnóstico de cáncer de mama que sobreexprese el gen HER2, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento Trastuzumab para el tratamiento farmacológico de cáncer de mama que sobreexprese el gen HER2.
c) Garantías de oportunidad
c.1. Tratamiento
En personas que tengan confirmación del diagnóstico de cáncer de mamas que sobreexprese el gen HER2, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Trastuzumab, dentro de 20 días desde la solicitud del tratamiento.
Estando en tratamiento con Trastuzumab, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 20 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
10. TRATAMIENTO CON ETANERCEPT O ABATACEPT O ADALIMUMAB O GOLIMUMAB O TOCILIZUMAB O TOFACITINIB O RITUXIMAB O BARICITINIB O UPADACITINIB EN PERSONAS CON ARTRITIS REUMATOIDE ACTIVA REFRACTARIA A TRATAMIENTO HABITUAL
La Artritis Reumatoide es una enfermedad inflamatoria crónica, autoinmune, y sistémica, de etiología desconocida que afecta principalmente a las articulaciones, evolucionando frecuentemente hacia la destrucción y deformidad articular. Se caracteriza por inflamación poliarticular y simétrica de pequeñas y grandes articulaciones, con posible compromiso sistémico extraarticular en cualquier momento de su evolución. Las personas experimentan dolor crónico y discapacidad progresiva.
a) Población beneficiaria
Personas con diagnóstico de artritis reumatoide activa refractaria a tratamiento habitual que cumplan los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1 Tratamiento
Medicamento Etanercept o Abatacept o Adalimumab o Golimumab o Tocilizumab o Tofacitinib o Rituximab o Baricitinib o Upadacitinib, para el tratamiento de artritis reumatoide activa refractaria a tratamiento habitual.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico de artritis reumatoide activa refractaria a tratamiento habitual que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Etanercept o Abatacept o Adalimumab o Golimumab o Tocilizumab o Tofacitinib o Rituximab o Baricitinib o Upadacitinib, según la indicación del médico tratante registrada al momento de generar la solicitud, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
11. PROFILAXIS DE LA INFECCIÓN DEL VIRUS RESPIRATORIO SINCICIAL CON PALIVIZUMAB PARA PREMATUROS MENORES DE 32 SEMANAS Y LACTANTES MENORES DE 1 AÑO CON CARDIOPATÍAS CONGÉNITAS HEMODINÁMICAMENTE SIGNIFICATIVAS
La infección por Virus Respiratorio Sincicial (VRS) es una causa viral principal de infección aguda de las vías respiratorias inferiores en lactantes y niños pequeños. Actualmente no existe cura para la infección por VRS y el tratamiento es principalmente de apoyo. Por lo tanto, la prevención es muy importante. Palivizumab es un anticuerpo monoclonal humanizado, dirigido contra el sitio antigénico A en la proteína F del VRS. Tiene una actividad inhibitoria de la fusión y es un potente neutralizante frente al subtipo A y cepas B del virus.
a) Población beneficiaria
En prematuros(as) menores de 32 semanas de edad gestacional al nacer o menos de 1.500 g de peso al nacer y su hermano gemelo, y que al inicio del período de alta circulación viral tengan menos de 1 año de edad cronológica.
En lactantes con cardiopatías congénitas hemodinámicamente significativas no resueltas o cardiopatía cianótica secundaria a cardiopatía de alta complejidad, y que al inicio del período de alta circulación viral tengan menos de 1 año de edad cronológica.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento palivizumab para profilaxis de infección de virus respiratorio sincicial.
c) Garantía de oportunidad:
c.1. Tratamiento
Se realizará la administración de una dosis mensual de profilaxis con palivizumab, durante el periodo de máxima circulación viral con un máximo de 5 dosis anuales, en aquellos pacientes que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud:
En recién nacidos prematuros de menos de 32 semanas de edad gestacional al nacer o menos de 1500 grs de peso al nacimiento y su hermano gemelo, que al inicio del periodo de máxima circulación viral tengan menos de 1 año de edad cronológica, el inicio del tratamiento podrá realizarse. 24 horas después del nacimiento o 24 horas después de la extubación en pacientes que estuvieron en ventilación mecánica, y al menos 72 horas previas al alta o de forma ambulatoria, si ya se encuentran en su domicilio. En recién nacidos o lactantes con cardiopatía congénita hemodinámicamente significativa no resuelta o cardiopatía secundaria cianótica a cardiopatía de alta complejidad, que al inicio del periodo de máxima circulación viral tengan menos de 1 año de edad cronológica, el inicio del tratamiento podrá realizarse 24 horas después del nacimiento o 24 horas después de la extubación en pacientes que estuvieron en ventilación mecánica, y al menos 72 horas previas al alta o de forma ambulatoria, si ya se encuentran en su domicilio. Estando en tratamiento con palivizumab, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo a lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud. Dichas prestaciones serán cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
12. TRATAMIENTO CON INFLIXIMAB O ADALIMUMAB PARA PERSONAS CON ENFERMEDAD DE CROHN GRAVE REFRACTARIA A TRATAMIENTO HABITUAL
La Enfermedad de Crohn (EC) forma parte del grupo de enfermedades inflamatorias intestinales que puede afectar a cualquier parte del tubo digestivo y tiende a tener un compromiso segmentario. Las áreas que se comprometen con mayor frecuencia son el íleon terminal y el ciego. Esta enfermedad se caracteriza por episodios de actividad y remisión de la inflamación, de curso progresivo que puede avanzar a la estenosis o formación de fístulas.
a) Población beneficiaria
Personas con diagnóstico de enfermedad de Crohn grave refractaria a tratamiento habitual, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento Adalimumab o Infliximab, para el tratamiento de enfermedad de Crohn grave refractaria a tratamiento habitual.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico de enfermedad de Crohn grave, ante el fracaso al tratamiento habitual con medicamentos de primera línea (glucocorticoides, inmunosupresores), que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con adalimumab o Infliximab, según la indicación del médico tratante registrada al momento de generar la solicitud, de acuerdo a:
- Para pacientes con enfermedad de Crohn grave hospitalizados: En un plazo de 7 días desde la solicitud del tratamiento. Tendrá derecho a continuación de la inducción en un plazo de 10 días, desde la solicitud del tratamiento.
- Para pacientes con enfermedad de Crohn grave no hospitalizados: En un plazo de 30 días, desde la solicitud del tratamiento.
- Para pacientes con enfermedad de Crohn con fístulas perianales complejas: En un plazo de 30 días desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 30 días desde la solicitud del tratamiento y en caso de estar hospitalizado dentro de 7 días, desde la solicitud del tratamiento.
a) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
13. NUTRICIÓN ENTERAL DOMICILIARIA TOTAL O PARCIAL, PARA PERSONAS CUYA CONDICIÓN DE SALUD IMPOSIBILITA LA ALIMENTACIÓN POR VÍA ORAL
La Nutrición Enteral (NE) es una técnica de soporte nutricional mediante la cual se aportan sustancias nutritivas directamente al aparato digestivo, por medio de sondas instaladas por vía nasal u ostomía en pacientes que por su condición de salud no pueden cubrir sus requerimientos por vía oral o esta vía está contraindicada, pero cuentan con tracto gastrointestinal con suficiente capacidad funcional. La Nutrición Enteral Domiciliaria (NED) está indicada en pacientes clínicamente estables, con el objetivo de garantizar el tratamiento nutricional y los cuidados correspondientes en un entorno más cómodo para el paciente.
a) Población beneficiaria
Personas cuya condición de salud imposibilita la alimentación por vía oral. Para efectos de este decreto, se considerará que una persona está imposibilitada para alimentarse por vía oral, cuando requiera al menos un 30% del aporte nutricional por vía enteral.
b) Prestaciones garantizadas
b.1. Tratamiento
b.1.1. Fórmulas de alimentación enteral
Fórmulas poliméricas, oligoméricas, elementales (monoméricas) o especiales, según el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b.1.2. Dispositivos de uso médico
Dispositivos de uso médicos necesarios para alimentación enteral domiciliaria vía sonda nasogástrica, sonda nasoyeyunal, ostomía gástrica u ostomía yeyunal, y sus recambios, renovaciones o mantenciones, según lo definido en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
Sonda Nasogástrica
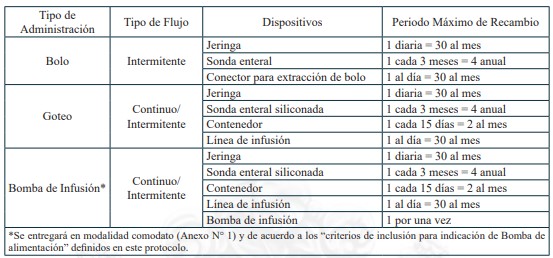
Sonda Nasoyeyunal
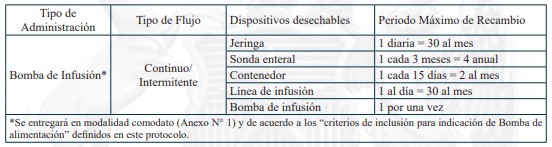
Ostomía gástrica con botón de ostomía
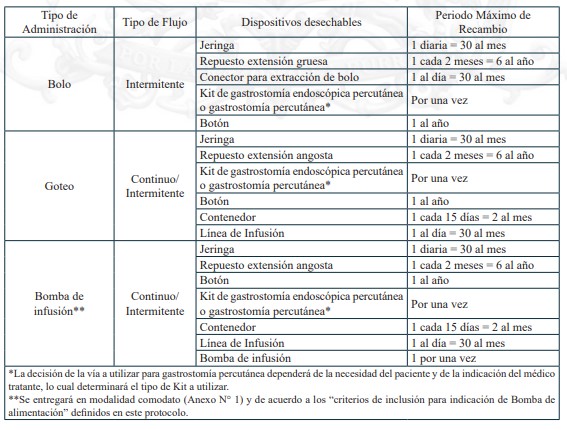
Ostomía gástrica con sonda gastrostomía
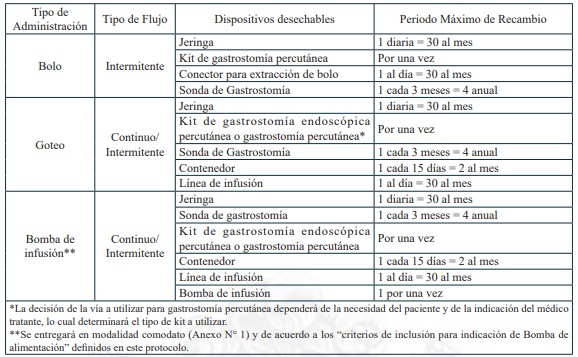
Ostomía yeyunal con botón de ostomía
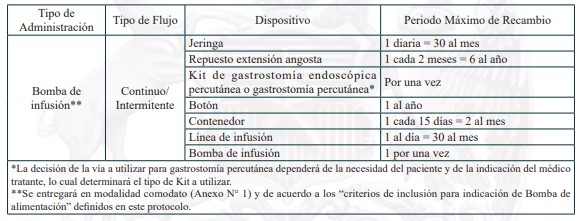
Ostomía yeyunal con sonda de yeyunostomía
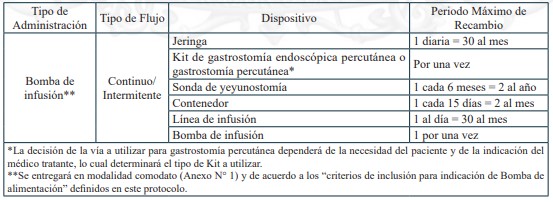
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con confirmación diagnóstica del requerimiento de nutrición enteral domiciliaria, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se les hará entrega de los alimentos y dispositivos médicos para la nutrición enteral domiciliaria, asignados al beneficiario, en un plazo no mayor a 30 días, desde la solicitud del tratamiento.
En caso de estar recibiendo las fórmulas y dispositivos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuar con su recepción. El inicio se realizará dentro de 30 días desde la solicitud del tratamiento.
a) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
14. TRATAMIENTO BASADO EN LA ADMINISTRACIÓN DE INSULINA, A TRAVÉS DE INFUSORES SUBCUTÁNEOS DE INSULINA (BOMBAS DE INSULINA) PARA PERSONAS CON DIAGNÓSTICO DE DIABETES MELLITUS TIPO 1 INESTABLE SEVERA
La diabetes tipo 1 (DM1) inestable severa, corresponde a aquella DM 1 que cursa con alta variabilidad glicémica (inestabilidad) y que conlleva a complicaciones severas y recurrentes, como la cetosis y cetoacidosis diabética, emergencia metabólica aguda potencialmente mortal, desencadenada por la hiperglicemia, así como eventos de hipoglicemia severa inadvertida cuya gravedad pueden conllevar desde convulsiones, el coma y la muerte.
a) Población beneficiaria
Personas con sospecha o diagnóstico de diabetes mellitus tipo 1 inestable o severa, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1 Confirmación diagnóstica
Monitoreo continuo de glicemia.
b.2 Tratamiento
b.2.1 Dispositivo de uso médico
Infusor subcutáneo continuo de insulina con sensor, junto a sus insumos, de acuerdo a los subgrupos establecidos en el protocolo específico para esta condición de salud.
c) Garantías de oportunidad
c.1. Tratamiento
En personas con diagnóstico de diabetes mellitus tipo 1, inestable severa que cumpla con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se entregará al beneficiario el dispositivo de uso médico infusor subcutáneo continuo de insulina (bomba de insulina) con sensor de glicemia, junto a sus insumos, dentro de 60 días desde la solicitud y confirmación del tratamiento.
Los recambios, las renovaciones o las mantenciones de los dispositivos médicos necesarios señalados en la letra anterior durante el período de tratamiento.
En caso de disponer del infusor subcutáneo continuo de insulina con sensor, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso al recambio o renovación, según lo definido de forma previa.
d) Continuidad de la atención.
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
15. DISPOSITIVO DE ESTIMULACIÓN CEREBRAL PROFUNDA PARA PERSONAS CON DISTONÍA GENERALIZADA
La distonía es un trastorno del movimiento caracterizado por contracciones musculares sostenidas o intermitentes que causan posturas o movimientos anormales, a menudo repetitivos, o ambos. Los movimientos distónicos generalmente son modelados, retorcidos y pueden ser temblorosos. La distonía a menudo se inicia o empeora por acción voluntaria y se asocia con el desbordamiento de la activación muscular.
Dentro de las clasificaciones de distonías se encuentra aquella según la distribución en el cuerpo que esté afectada, en donde se identifica la de tipo generalizada, la cual involucra el tronco y al menos 2 otras partes del cuerpo.
a) Población beneficiaria
Personas con diagnóstico de distonía generalizada que cumplan los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
b.2.1 Dispositivo de uso médico
Dispositivo de estimulación cerebral profunda: generador de pulsos implantable, extensión y electrodos.
Generador de pulsos implantable de reemplazo para dispositivo de estimulación cerebral profunda.
c) Garantías de oportunidad
c.1. Tratamiento
Para personas con diagnóstico de distonía generalizada, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se entregará al prestador aprobado en la etapa de tratamiento, el dispositivo de estimulación cerebral asignado al beneficiario en un plazo de 30 días desde la solicitud del tratamiento.
La entrega al prestador aprobado en la etapa de tratamiento del "generador de pulsos implantable" de reemplazo, en personas que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se realizará considerando un mínimo de 150 días antes de la fecha de certificación de vida útil del generador de pulsos implantables informada por el proveedor. Será responsabilidad del médico tratante realizar el registro del requerimiento en las vías definidas para ello.
En caso de disponer del dispositivo de estimulación cerebral profunda, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a la entrega del "generador de pulsos implantable" de reemplazo compatible, según lo indicado de forma previa.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
16. TRATAMIENTO CON SUNITINIB O EVEROLIMUS PARA PERSONAS CON ENFERMEDAD PROGRESIVA DE TUMORES NEUROENDOCRINOS PANCREÁTICOS
Los tumores neuroendocrinos son neoplasias provenientes del sistema neuroendocrino y están integrados por células productoras de aminas y de ácidos con diferentes perfiles hormonales según el sitio de origen. Las células neuroendocrinas están ampliamente distribuidas a través del cuerpo, por lo que estas neoplasias se pueden presentar en la mayoría de los órganos. Los tumores neuroendocrinos se dividen según su origen en aquellos provenientes del tubo digestivo, tumores neuroendocrinos pancreáticos, tumores neuroendocrinos torácicos o pulmonares y de otras ubicaciones. Los tumores neuroendocrinos pancreáticos se originan en las células de los islotes de Langerhans. Pueden ser funcionales o no funcionales.
a) Población beneficiaria
Personas mayores de 18 años con diagnóstico de tumores neuroendocrinos pancreáticos progresivos y bien diferenciados con enfermedad irresecable, localmente avanzada o metastásica, que cumplan los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamentos Sunitinib o Everolimus, para el tratamiento de enfermedad progresiva de tumores neuroendocrinos pancreáticos.
c) Garantías de oportunidad
c.1 Tratamiento
Las personas mayores de 18 años con diagnóstico de tumores neuroendocrinos pancreáticos progresivos y bien diferenciados con enfermedad irresecable, localmente avanzada o metastásica, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciaran el tratamiento con Sunitinib o Everolimus, según la indicación del médico tratante, en un plazo de 15 días desde la recepción de la solicitud del medicamento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 15 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
17. DISPOSITIVO DE IMPLANTE COCLEAR UNILATERAL PARA PERSONAS CON HIPOACUSIA SENSORIONEURAL BILATERAL SEVERA O PROFUNDA POSTLOCUTIVA
El término compuesto "Hipoacusia Sensorioneural Severa o Profunda" hace referencia, por una parte, a una condición audiológica de disminución, desde 70 dB - 90 dB (Severa y Profunda, respectivamente), en la capacidad auditiva y, por otra, a la localización coclear o retrococlear (nervio auditivo) de un daño o lesión, uni o bilateral.
a) Población beneficiaria
Personas mayores de 4 años con diagnóstico de hipoacusia sensorioneural bilateral severa o profunda postlocutivas, que cumplan los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud y con indicación médica de uso del dispositivo de implante coclear y el recambio de sus accesorios.
b) Prestaciones garantizadas
b.1 Dispositivo de uso médico
Dispositivo de implante coclear unilateral, sus accesorios y el recambio de estos.
c) Garantías de oportunidad
c.1. Dispositivo de uso médico
En personas con diagnóstico de hipoacusia sensorioneural bilateral severa o profunda, postlocutiva desde los 4 años, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se entregará al prestador aprobado para la etapa de tratamiento el dispositivo asignado al beneficiario, dentro de 45 días desde la solicitud del tratamiento.
En personas con diagnóstico de hipoacusia post meningitis o hipoacusia autoinmune bilateral, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, se entregará al prestador aprobado en la etapa de tratamiento el dispositivo asignado al beneficiario dentro de 15 días desde la solicitud del tratamiento.
El recambio de accesorios de implante coclear se realizará en aquellas personas que cumplan los criterios de inclusión y de acuerdo con la periodicidad establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud dependiendo del tipo de accesorio. Será responsabilidad del médico tratante realizar el registro del requerimiento en las vías definidas para ello.
El reemplazo de procesador del habla se realizará en aquellas personas que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, cada 5 años desde la instalación del dispositivo de implante coclear o el último recambio. Será responsabilidad del médico tratante realizar el registro del requerimiento en las vías definidas para ello.
En caso de disponer del dispositivo de implante coclear, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso al recambio de accesorios y reemplazo del procesador del habla compatible, según lo indicado de forma previa.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
18. TRATAMIENTO CON INHIBIDOR DE C1 ESTERASA PARA PERSONAS CON ANGIOEDEMA HEREDITARIO
El Angioedema es una enfermedad infrecuente que se define como una reacción vascular de la dermis profunda o de los tejidos subcutáneos/submucosos con dilatación localizada y aumento de la permeabilidad de los vasos sanguíneos, que produce inflamación del tejido. Existe el subtipo de angioedema hereditario, el cual se debe a deficiencias del inhibidor de C1. Este se transmite en un patrón genético autosómico dominante que causa una gran variedad de mutaciones diferentes del gen SERPING1 las que dan como resultado la deficiencia del inhibidor de C1 y una regulación alterada de la síntesis de bradiquinina, y por lo tanto una inhibición deficitaria del sistema complemento (fundamental en la respuesta inmunitaria).
a) Población beneficiaria
Personas con diagnóstico de angioedema hereditario de tipo I o II, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Inhibidor de C1 esterasa ante episodio agudo de angioedema hereditario con deficiencia de inhibidor de C1.
Inhibidor de C1 esterasa como profilaxis a corto plazo en caso de cirugía mayor de cabeza, cuello u oral.
c) Garantías de oportunidad
c.1. Tratamiento
Ante episodio agudo de angioedema hereditario, según indicación médica recibirán inhibidor de C1 esterasa, de forma inmediata en servicio de urgencia de la red de prestadores aprobados en la etapa de tratamiento (nivel de priorización C2 o ESI2) (1)
Como tratamiento de profilaxis a corto plazo en cirugía mayor de cabeza, cuello u oral, se deberá administrar como máximo con 2 horas de anticipación a la cirugía.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
19. AYUDAS TÉCNICAS PARA PERSONAS CON ESCLEROSIS LATERAL AMIOTRÓFICA MODERADA O SEVERA
La Esclerosis Lateral Amiotrófica (ELA) es un desorden neurodegenerativo, constituyendo un subgrupo dentro de las enfermedades neuromusculares motoras, las cuales son un grupo de patologías que afectan al sistema nervioso periférico, en alguno de los componentes de la unidad motora. No posee una cura conocida, causando debilidad muscular progresiva, discapacidad y eventualmente la muerte. Las ayudas técnicas favorecen el logro de los objetivos terapéuticos de rehabilitación.
a) Población beneficiaria
Personas con diagnóstico de esclerosis lateral amiotrófica moderada o severa que requieran ayudas técnicas y cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
b.1.1 Ayuda técnicas
Ayudas técnicas para personas con Esclerosis Lateral Amiotrófica moderada y severa, según protocolo específico para esta condición de salud.
b.1.1. Ayudas técnicas para el desempeño de las actividades de la vida diaria (AVD):
a) Baño portátil.
b) Silla de ruedas neurológica.
c) Tecnologías de la comunicación aumentativas y alternativas (tecnologías de seguimiento ocular).
b.1.2. Ayudas técnicas para el tratamiento rehabilitador:
a) Colchón antiescaras.
b) Cojín antiescaras.
b.1.3. Ayudas técnicas para el soporte vital:
a) Equipo ventilador mecánico no invasivo domiciliario con generador de flujo a presión positiva binivelada (Bi-PAP).
b) Aspirador de secreciones.
c) Garantías de oportunidad
____________________
(1) Ordinario C2/N° 313: Informa sobre herramienta ESI en la UEH del país, Subsecretaría de Redes Asistenciales, 26 de enero del 2018.
c.1. Tratamiento
En personas con diagnóstico de esclerosis lateral amiotrófica que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, recibirán las ayudas técnicas asignadas al beneficiario, en un plazo de 30 días, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
20. DISPOSITIVOS DE USO MÉDICO PARA CURACIONES EN PERSONAS CON EPIDERMÓLISIS BULOSA DISTRÓFICA O DE LA UNIÓN (JUNTURAL)
La epidermólisis bulosa es un grupo de desórdenes hereditarios, caracterizados por la excesiva susceptibilidad de la piel y mucosas a separarse de su tejido subyacente luego de un trauma mecánico. Es causada por mutaciones que afectan a las proteínas estructurales de la piel, pudiendo afectar también mucosas, como las de la cavidad oral, esófago, cavidad nasal, faringe, tracto genitourinario, zona perianal y conjuntiva. No existe un tratamiento específico y su evolución es crónica. La rehabilitación es el mecanismo para lograr una mantención favorable de la enfermedad respecto a la movilización y realización de actividades de la vida diaria.
a) Población beneficiaria
Recién Nacidos con sospecha diagnóstica de epidermólisis bulosa distrófica o de la unión y personas con diagnóstico de epidermólisis bulosa distrófica o de la unión, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
b.1.1 Dispositivo de uso médico
Dispositivos de uso médico para curaciones para personas con epidermólisis bulosa distrófica o de la unión según lo definido en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
Dispositivos de uso médico para curaciones:
1. Kit de curación desechable.
2. Vendas de gasa elástica.
3. Vendaje tubular de contención.
4. Gasas no tejidas.
5. Gasa absorbente en rollo.
6. Agujas hipodérmicas.
7. Apósito de contacto flexible con tecnología lípido coloide (TLC).
8. Solución con agua purificada, undecilenamidopropil betaína y polihexanida, para el lavado, descontaminación e hidratación de heridas.
9. Gel altamente viscoso compuesto por glicerol, agua purificada, undecilenamidopropil betaína, polihexanida, hidroxietilcelulosa, para el lavado, descontaminación e hidratación de heridas.
10. Rollo de gasa oclusiva con Tibromofenato de bismuto al 3% en una mezcla con petrolato.
11. Hidrogel amorfo, translúcido e incoloro.
12. Apósito absorbente, extrafino y autoadherente con tecnología de adhesivos con silicona.
13. Apósito absorbente.
14. Apósito de espuma (espuma de poliuretano y tecnología de adhesivos de silicona).
15. Apósito Interfase de contacto flexible, antibacteriana con tecnología lípido coloide (TLC) y plata.
16. Apósito de espuma microadherente con tecnología lípido coloide (TLC) y factor nano oligosacárido (NOSF).
17. Apósito absorbente antimicrobiano y plata iónica.
18. Apósito de espuma de hidrofibra.
19. Apósito hidroconductivo.
20. Apósito hidroactivo con poliacrilato superabsorbente (SAP).
21. Apósito de malla de acetato de celulosa y petrolato.
22. Apósito de gasa parafinada de baja adherencia.
23. Apósito de transferencia de exudado.
24. Apósito de membrana polimérica multifuncional.
25. Apósito antimicrobiano de espuma de poliuretano absorbente con plata y capa de silicona.
26. Apósito de hidrofibra de hidrocoloide con fibra reforzante de celulosa.
27. Cinta quirúrgica de rayón altamente respirable, no oclusiva y con adhesivo hipoalergénico.
28. Cinta de fijación de silicona atraumática
c) Garantías de oportunidad
c.1. Tratamiento
En recién nacidos con sospecha diagnóstica de epidermólisis bulosa distrófica o de la unión, y que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, recibirán los dispositivos de uso médico para curaciones asignados al beneficiario, en un plazo de 24 horas, desde la solicitud del tratamiento.
En personas hospitalizadas, cursando una infección o con indicación de cirugía que tengan diagnóstico confirmado de epidermólisis bulosa distrófica o de la unión y que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, recibirán los dispositivos de uso médico para curaciones asignados al beneficiario, en un plazo de 48 horas, desde la solicitud del tratamiento.
En personas con diagnóstico confirmado de epidermólisis bulosa distrófica o de la unión que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, recibirán los dispositivos de uso médico para curaciones asignados al beneficiario, en un plazo de 30 días, desde la solicitud del tratamiento.
d) Continuidad de tratamiento
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
21. TRATAMIENTO CON IMATINIB O SUNITINIB EN PERSONAS CON TUMORES DEL ESTROMA GASTROINTESTINAL NO RESECABLES O METASTÁSICOS
Los tumores del estroma gastrointestinal (GIST) son un grupo de neoplasias que representa menos del 1% de los tumores primarios del tracto gastrointestinal. Se localizan preferentemente en el estómago y el intestino delgado, aunque pueden desarrollarse en cualquier lugar del aparato digestivo e incluso fuera de él.
a) Población beneficiaria
Personas con sospecha o diagnóstico de tumor del estroma gastrointestinal no resecable o metastásico que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Examen inmunohistoquímico de proteína c-kit/CD117 y tomografía computada.
b.2. Tratamiento
Medicamento Imatinib o Sunitinib para tratamiento de tumores del estroma gastrointestinal (GIST).
c) Garantías de oportunidad
c.1. Confirmación diagnóstica
Las personas con sospecha clínica fundada de tumores del estroma gastrointestinal no resecable o metastásico tendrán acceso a examen de inmunohistoquímica de proteína c-kit/CD117 y tomografía computada, en un plazo de 30 días desde la recepción del formulario de sospecha fundada por el prestador aprobado para la etapa de confirmación.
c.2. Tratamiento
Las personas con diagnóstico confirmado de tumores del estroma gastrointestinal no resecable o metastásico, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con imatinib, en un plazo de 20 días, desde la solicitud del tratamiento.
En aquellas personas que estando en tratamiento con Imanitib progresen en su condición y que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Sunitinib, en un plazo de 20 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 20 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
22. TRATAMIENTO CON GOLIMUMAB O ETANERCEPT O ADALIMUMAB O SECUKINUMAB EN PERSONAS CON ARTRITIS PSORIÁSICA MODERADA A GRAVE REFRACTARIA A TRATAMIENTO HABITUAL
La artritis psoriásica (APs) es una enfermedad inflamatoria crónica del sistema musculoesquelético generalmente asociada a psoriasis, que pertenece al grupo de las Espondiloartritis. Tiene diversas presentaciones clínicas, pudiendo afectar la columna vertebral y articulaciones sacroilíacas, las articulaciones periféricas, las entesis, las vainas tendinosas, las uñas y otros órganos como el intestino o el ojo.
a) Población beneficiaria
Personas con diagnóstico de artritis psoriásica moderada a grave refractaria al tratamiento habitual, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamentos Golimumab o Etanercept o adalimumab o Secukinumab, para el tratamiento de artritis psoriásica moderada a grave refractaria al tratamiento habitual.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico confirmado de artritis psoriásica moderada a grave refractaria al tratamiento habitual, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Golimumab o Etanercept o Adalimumab o Secukinumab, según la indicación del médico tratante registrada al momento de generar la solicitud, en un plazo de 60 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
23. TRATAMIENTO CON GOLIMUMAB O ADALIMUMAB PARA PERSONAS CON COLITIS ULCEROSA MODERADA E INFLIXIMAB EN PERSONAS CON COLITIS ULCEROSA GRAVE, REFRACTARIA AL TRATAMIENTO DE PRIMERA LÍNEA.
La colitis ulcerosa es una enfermedad inflamatoria crónica que afecta la mucosa del colon en forma continua, comprometiendo el recto y una porción variable de la extensión del resto del colon, sin la presencia de granulomas. Los pacientes con colitis ulcerosa usualmente presentan diarrea, sangrado rectal, dolor abdominal, tenesmo e incontinencia. Las causas de esta enfermedad no son conocidas, aunque se puede atribuir un mayor riesgo en personas con antecedentes familiares.
a) Población beneficiaria
Personas con diagnóstico de colitis ulcerosa moderada y grave refractaria a tratamiento de primera línea, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de garantías de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
b.1.1 Medicamento golimumab o adalimumab en personas adultas con diagnóstico confirmado de colitis ulcerosa moderada inmunorefractaria.
b.1.2 Medicamento infliximab en personas adultas con diagnóstico confirmado de colitis ulcerosa grave refractaria a corticoides.
b.1.3 Medicamento infliximab en pacientes pediátricos con diagnóstico confirmado de colitis ulcerosa grave refractaria a corticoides o colitis ulcerosa moderada inmunorefractaria.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico de colitis ulcerosa moderada y grave refractaria a tratamiento de primera línea, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento, según la indicación del médico tratante registrada al momento de generar la solicitud, de acuerdo con lo siguiente:
c.1.1 En personas adultas y pediátricas con diagnóstico de colitis ulcerosa grave, hospitalizados con crisis cortico-refractaria, recibirán tratamiento con Infliximab en un plazo no mayor a 3 días, desde la solicitud del tratamiento.
c.1.2 En personas adultas con diagnóstico de colitis ulcerosa moderada inmunorefractaria, recibirán tratamiento con Golimumab o Adalimumab, en un plazo de 30 días, desde la solicitud del tratamiento.
c.1.3 En pacientes pediátricos con diagnóstico de colitis ulcerosa moderada inmunorefractaria, recibirán tratamiento con Infliximab en un plazo de 30 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 30 días desde la solicitud del tratamiento y en caso de estar hospitalizado con crisis cortico-refractaria, dentro de 3 días, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
24. TRATAMIENTO CON TETRABENAZINA PARA LA COREA EN PERSONAS CON ENFERMEDAD DE HUNTINGTON
La enfermedad de Huntington es un trastorno neurodegenerativo heredado progresivo y fatal caracterizado por disfunción motora, cognitiva y del comportamiento, todo lo cual contribuye a la discapacidad acumulativa y la pérdida de la calidad de vida. En la actualidad no existe una cura para esta enfermedad y el tratamiento está enfocado en disminuir la sintomatología motora de este problema de salud.
a) Población beneficiaria
Personas con sospecha o diagnóstico confirmado de enfermedad de Huntington, que presentan corea y que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Confirmación diagnóstica
Test genético de repetición de CAG del exón 1 del Gen HTT.
b.2. Tratamiento
Medicamento Tetrabenazina, para el tratamiento de la corea en enfermedad de Huntington.
c) Garantías de oportunidad
c.1. Confirmación diagnóstica
Las personas con sospecha de enfermedad de Huntington y corea, se realizará el test de repetición de CAG del exón 1 del Gen HTT, dentro de 30 días, desde la recepción del formulario de sospecha fundada y la muestra de sangre venosa, por el prestador aprobado en la etapa de confirmación.
c.2. Tratamiento
El inicio del tratamiento con Tetrabenazina, se realizará en personas con corea y diagnóstico confirmado de enfermedad de Huntington, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones, dentro de 60 días desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con Tetrabenazina, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
25. TRATAMIENTO CON INMUNOGLOBULINA G EN PERSONAS CON INMUNODEFICIENCIAS PRIMARIAS
Las inmunodeficiencias primarias conforman un grupo heterogéneo de más de 340 diferentes enfermedades de origen monogénico. Estas enfermedades se caracterizan por comprometer la función normal de las distintas ramas del sistema inmune y por manifestarse mediante una combinación de infecciones recurrentes, trastornos autoinmunes, trastornos autoinflamatorios, trastornos linfoproliferativos, procesos granulomatosos, enfermedades alérgicas graves y malignidad.
a) Población beneficiaria
Personas con diagnóstico de inmunodeficiencia primaria, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento inmunoglobulina G endovenosa o inmunoglobulina G subcutánea para el tratamiento de la inmunodeficiencia primaria.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico de inmunodeficiencia primaria, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento, con inmunoglobulina G endovenosa en un plazo de 14 días, desde la solicitud del tratamiento.
Las personas con diagnóstico de inmunodeficiencia primaria, que cumplan con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y que presenten al menos una de las siguientes condiciones: anafilaxia documentada debida a inmunoglobulina G endovenosa síntomas documentados relacionados a la infusión de inmunoglobulina G endovenosa no manejables o imposibilidad certificada de obtener accesos venosos, iniciarán el tratamiento, con inmunoglobulina G subcutánea en un plazo de 14 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con alguno de los medicamentos garantizados, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 14 días desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
26. TRATAMIENTO CON BELIMUMAB PARA PERSONAS CON LUPUS ERITEMATOSO SISTÉMICO CON COMPROMISO CUTÁNEO O ARTICULAR REFRACTARIO A TRATAMIENTO HABITUAL
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune inflamatoria crónica de causa desconocida que puede afectar cualquier órgano. Se caracteriza por un curso cíclico con remisiones y recaídas. Puede causar una discapacidad física y funcional importante y sus manifestaciones varían desde una afectación cutánea y articular relativamente leve, hasta una fatiga debilitante, un deterioro cognitivo significativo, enfermedad renal en etapa terminal y trombosis.
a) Población beneficiaria
Personas con diagnóstico de lupus eritematoso sistémico con compromiso cutáneo o articular refractario a tratamiento habitual y que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento Belimumab para el tratamiento de lupus eritematoso sistémico con compromiso cutáneo o articular refractario a tratamiento habitual.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico confirmado de lupus eritematoso sistémico con compromiso cutáneo o articular refractario a tratamiento habitual, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Belimumab, en un plazo de 60 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con Belimumab, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
27. TRATAMIENTO CON RUXOLITINIB PARA PERSONAS CON MIELOFIBROSIS PRIMARIA Y SECUNDARIA A OTRAS NEOPLASIAS MIELOPROLIFERATIVAS
La Mielofibrosis es una neoplasia mieloproliferativa crónica, generalmente idiopática, caracterizada por la proliferación desregulada de células mieloides (incluyendo megacariocitos y progenitores mieloides y eritroides) en la médula ósea, de variable madurez morfológica y eficacia hematopoyética. Frecuentemente se presenta de manera asintomática, siendo motivos frecuentes de consulta la esplenomegalia, hepatomegalia, o hemograma alterado.
a) Población beneficiaria
Personas con diagnóstico de Mielofibrosis primaria o Mielofibrosis secundaria a otras neoplasias mieloproliferativas, que cumplan con los criterios establecidos en el protocolo para el otorgamiento de las prestaciones de este problema de salud.
b) Prestaciones garantizadas
b.1. Tratamiento
Medicamento Ruxolitinib, para el tratamiento de Mielofibrosis primaria o Mielofibrosis secundaria a otras neoplasias mieloproliferativas.
c) Garantías de oportunidad
c.1. Tratamiento
Las personas con diagnóstico de Mielofibrosis primaria o Mielofibrosis secundaria a otras neoplasias mieloproliferativas, que cumplan los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, iniciarán el tratamiento con Ruxolitinib, en un plazo de 60 días, desde la solicitud del tratamiento.
Estando en tratamiento farmacológico con Ruxolitinib, previo a ser beneficiario de esta ley, y de cumplir con los criterios determinados en el protocolo para el otorgamiento de las prestaciones de este problema de salud, tendrá acceso a continuarlo. El inicio se realizará dentro de 60 días, desde la solicitud del tratamiento.
d) Continuidad de la atención
La continuidad de la atención y controles clínicos posteriores, se realizarán de acuerdo con lo establecido en el protocolo para el otorgamiento de las prestaciones de este problema de salud, y las consultas médicas y exámenes complementarios si fuesen necesarios, deberán ser cubiertas por los seguros de salud correspondientes, de acuerdo con el plan de salud del beneficiario(a).
2º Déjase establecido que los tratamientos garantizados conforme a lo dispuesto en el presente decreto, se otorgarán exclusivamente de acuerdo a los protocolos aprobados para cada uno de los tratamientos asociados a enfermedades o condiciones de salud especificas a través de las resoluciones exentas Nº 735, de 26 de octubre de 2015, 1.447, de 28 de noviembre de 2016, 1.664, de 29 de diciembre de 2017, y 38, de 17 de enero de 2019, todas del Ministerio de Salud. De este modo, en los casos en que se aprueba más de una alternativa terapéutica para un tratamiento asociado a una condición de salud determinada, la indicación del medicamento específico se efectuará conforme al protocolo, según las condiciones clínicas del paciente.
3º Déjase establecido que las prestaciones a que tienen derecho los beneficiarios se encuentran taxativamente señaladas en el presente decreto, las que se entregarán de acuerdo a lo dispuesto en el numeral 1 de la parte resolutiva, señalando la frecuencia de las prestaciones, sin que proceda la homologación de las mismas. Para estos efectos, se entenderá por homologación de prestaciones el reemplazo de ellas por otras que no se encuentran contempladas en los numerales precedentes o con especificaciones distintas a las exigidas.
4º Déjase establecido que el Sistema de Protección Financiera para diagnósticos y tratamientos de alto costo de la ley Nº 20.850, cubrirá los diagnósticos y tratamientos contemplados en este decreto, exclusivamente para las indicaciones que se detallan en él, entendiendo que pueden existir otras situaciones o problemas de salud en las cuales se utilicen estos medicamentos, pero para los cuales no están contemplados.
5º Déjase establecido que conforme a lo dispuesto en el inciso primero, del artículo 9º, de la ley Nº 20.850, la revisión de la decisión podrá ser realizada hasta la dictación del próximo decreto que determina los diagnósticos y tratamientos de alto costo con sistema de protección financiera. Los efectos de la revisión de la decisión, se aplicarán a partir del 1 de julio del año 2022, para todos los diagnósticos y tratamientos incorporados en este decreto.
6º Establécese que el presente decreto regirá a contar del 1 de julio de 2019 y hasta el 30 de junio de 2022.
7º Derógase, el decreto Nº 47, de 2017, del Ministerio de Salud, a contar del 1 de julio del año 2019.
Anótese, tómese razón y publíquese.- SEBASTIÁN PIÑERA ECHENIQUE, Presidente de la República.- Emilio Santelices Cuevas, Ministro de Salud.- Felipe Larraín Bascuñán, Ministro de Hacienda.
Transcribo para su conocimiento decreto afecto Nº 2, de 18-01-2019.- Saluda atentamente a Ud., Paula Daza Narbona, Subsecretaria de Salud Pública.