MODIFICA LOS REGISTROS SANITARIOS DE MEDICAMENTOS QUE CONTIENEN EL PRINCIPIO ACTIVO RANITIDINA Y ESTABLECE CONTROLES RESPECTO DE LA AUTORIZACIÓN DE USO Y DISPOSICIÓN DE DICHOS PRODUCTOS
Núm. 4.495 exenta.- Santiago, 28 de octubre de 2020.
Vistos estos antecedentes:
Las medidas adoptadas en 2019 por diversas agencias de medicamentos internacionales de referencia, tales como: Food and Drug Administration (FDA) de los Estados Unidos, la Agencia Europea de Medicamentos (EMA, por sus siglas en inglés), Health Canada y Therapeutic Good Administration (TGA) de Australia, sobre el retiro preventivo de medicamentos que contienen ranitidina en su composición, debido al hallazgo de impurezas de nitrosaminas, destacando la N-nitrosodimetilamina (NDMA); las recomendaciones entregadas por la Organización Mundial de la Salud (OMS) respecto de: las medidas normativas que pueden adoptar las agencias reguladoras de medicamentos, la toxicidad de las nitrosaminas, los métodos de ensayo y los límites de ingesta diaria permisible para estas impurezas; la decisión de la FDA, en abril de 2020, de retirar del mercado todos los productos farmacéuticos que contienen ranitidina, debido a la generación de impurezas en el producto terminado a través del tiempo, en condiciones normales de almacenamiento, pudiendo llegar a límites de exposición considerados inaceptables; la existencia de registros sanitarios vigentes en Chile, de productos farmacéuticos que contienen ranitidina como materia prima activa y su distribución a establecimientos asistenciales públicos, mediante Cenabast; y,
Considerando:
Primero: Que, la evidencia del potencial riesgo que puede generar la administración de nitrosaminas en forma prolongada en el tiempo a los pacientes, debido a sus propiedades carcinogénicas.
Segundo: Que, las nitrosaminas corresponden a una clase de compuestos nitrosados, que tienen la estructura química de un grupo nitroso unido a una amina (R1N(-R2)-N=O), y que han sido reconocidas como potentes agentes genotóxicos en varias especies animales, estando algunas clasificadas como probables o posibles carcinógenos en humanos por la Agencia Internacional para la Investigación del Cáncer (IARC) - grupo 2 A de la OMS / (IARC) - y grupo B2 de la EPA.
Tercero: Que, las impurezas de nitrosamina identificadas en ranitidina poseen una toxicidad significativa.
Cuarto: Que, existe incertidumbre por la eventual presencia de impurezas de nitrosaminas en la ranitidina, que, según se ha determinado, pueden generarse a lo largo de la vida útil del producto.
Quinto: Que, los compuestos nitrosados se encuentran clasificados como compuestos de "cohorte de preocupación" en la guía M7 (R1)(¹) "Evaluación y control de impurezas (mutagénicas) reactivas al ADN en productos farmacéuticos para limitar el potencial riesgo carcinogénico" de la Conferencia Internacional de Armonización (ICH), publicada en marzo de 2018, donde se indica que se debe realizar el control de cualquier carcinógeno mutagénico conocido (como los compuestos nitrosos) y asegurar límites máximos en los productos por debajo de un nivel que asegure un riesgo insignificante de cáncer humano asociado a la exposición de estas impurezas.
Sexto: Que, a la fecha, la FDA ha identificado impurezas de nitrosamina que teóricamente podrían estar presentes en productos farmacéuticos(²), tales como; (NDMA), N-nitrosodietilamina (NDEA), ácido N-nitroso-N-metil-4-aminobutanoico (NMBA), N-nitrosoisopropiletilamina (NIPEA), N-nitrosodiisopropilamina (NDIPA), N-nitrosodibutilamina (NDBA) y N-nitrosometilfenilamina (NMPA), de los cuales, en la práctica, seis se han detectado en sustancias farmacéuticas o productos farmacéuticos.
Séptimo: Que, aun cuando existe una serie de metodologías para establecer la Ingesta Aceptable (IA) de cada compuesto(3), desde el ámbito sanitario se ha escogido como representativa, la toxicidad de las nitrosaminas NDMA y NDEA. Es así que, durante el año 2018, la FDA estableció "Límites Provisionales" de IA, basados en la dosis media tóxica (TD50) en roedores, cuya extrapolación lineal se tradujo en la probabilidad de riesgo de cáncer de 1 en 100.000 durante una exposición crónica mayor a 70 años. La FDA ha utilizado estos Límites Provisionales para orientar la toma de decisiones balanceando los riesgos de una posible exposición a un carcinógeno a largo plazo con la interrupción del manejo clínico de los pacientes crónicos.
Octavo: Que, los Límites Provisionales establecidos por la Agencia de Alimentos y Drogas de Estados Unidos de América -FDA-, que sirven de base y orientación para la toma de decisiones sanitarias (ver Tabla Nº 1) son:
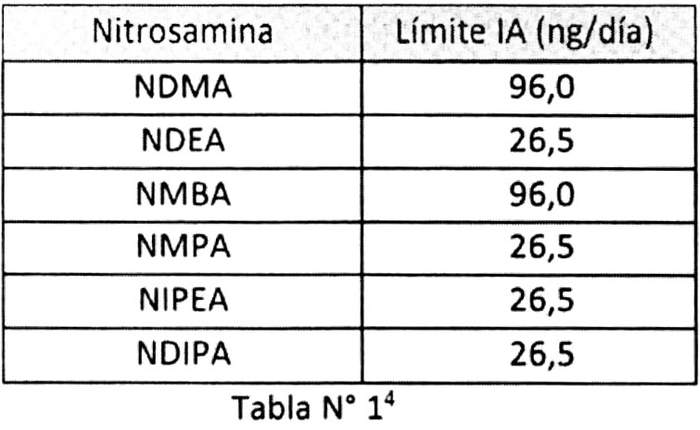
Noveno: Que, la conveniencia de adoptar los Límites Provisionales, en ausencia de información nacional respecto a toxicidad de nitrosaminas en particular, establecidos por la Agencia de Alimentos y Drogas de Estados Unidos de América -FDA- (Tabla Nº 1), y mantenerlas, mientras no se disponga de información suplementaria que sustente una decisión diferente.
Décimo: Que, en este contexto, descansa en la esfera de responsabilidad de esta autoridad, verificar que los productos farmacéuticos que sean distribuidos y comercializados en el país, cumplan con los requisitos de calidad, seguridad y eficacia que son exigibles al titular del registro, elementos que, a la luz de la normativa sanitaria vigente, son indispensables para la protección del bien jurídico salud pública.
------------------------
(1) International Council for Harmonisation of Technical Requirements of Pharmaceuticals for Human Use. ICH M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, 2017. https://www.ich.org/page/multidisciplinary-guidelines.
(2) Control of Nitrosamine Impurities in Human Drugs: Guidance for Industry. https://www.fda.gov/regulatory- information/search-fda-guidance-documents?utm_medium=email&utm_source=govdelivery.
(3) Control of Nitrosamine Impurities in Human Drugs: Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents?utm_medium=email&utm_source=govdelivery APPENDIX B.
(4) Nota: Estos límites son aplicables solo si un medicamento contiene una sola nitrosomina. Si se detecta más de una de las impurezas de nitrosamina identificadas en la Tabla 1 y la cantidad total de impurezas de nitrosomina excede 26,5 ng/día (IA para las nitrosaminas más potentes) basado en la dosis máxima diaria (MDD), el fabricante deberá comunicarse con este Instituto para su evaluación.
Se recomienda el uso de estos IA para determinar los límites de las impurezas de nitrosomina en los ingredientes activos y en los medicamentos.
Undécimo: Que, por tanto, deben establecerse lineamientos en relación a los productos farmacéuticos que contengan Ranitidina.
En este escenario, por una parte, el artículo 71 número 5 del DS número 3, de 2010, del Ministerio de Salud dispone: "El titular de registro sanitario es el responsable final de la seguridad y eficacia del medicamento.
Sin perjuicio de las obligaciones específicas establecidas en atención a la naturaleza de cada especialidad farmacéutica, todo titular de registro sanitario estará obligado a: 5) Mantener actualizado el registro sanitario, con arreglo al estado de la ciencia y la técnica, especialmente en relación a los métodos de control de calidad, así como a la seguridad y la eficacia de la especialidad farmacéutica".
En consecuencia, los titulares de registros sanitarios de productos que contienen el principio activo ranitidina, deberán actualizar la metodología analítica (farmacopea reconocida) mediante la cual se identificarán y cuantificarán las impurezas nitrosaminas y la realización de estudios de estabilidad en tiempo real, incorporando la determinación de impurezas de nitrosaminas, en los términos que se dispondrá en lo resolutivo del presente instrumento.
Duodécimo: Que, por otra parte, atendido que, tal como dispone el artículo 99 del DS Nº 3, de 2010, del Ministerio de Salud, que determina -como competencia de esta autoridad- el autorizar el uso y disposición de productos importados y sus materias primas, es fundamental que, en dicho procedimiento, se ejerza un control de parámetros asociados a la detección y cuantificación de impurezas.
En consecuencia, el solicitante deberá acreditar, para cada lote, mediante los certificados de análisis correspondientes que, la cantidad de impurezas detectada es inferior al límite máximo definido y que la cuantificación de impurezas nitrosaminas fue realizada mediante el método autorizado por este Instituto, en los términos que se dispondrá en lo resolutivo del presente instrumento.
Teniendo presente: Las disposiciones del artículo 96° del Código Sanitario; el Reglamento del Sistema Nacional de Control de Productos Farmacéuticos, aprobado por el decreto supremo Nº 3, de 2010, del Ministerio de Salud, los artículos 59° letra b) y 61° letra b), del DFL Nº 1, de 2005, y en uso de las facultades que me otorga el decreto Nº 51, de 2020, del Ministerio de Salud; así como lo establecido en la resolución Nº 7, de 2019, de la Contraloría General de la República, dicto la siguiente:
Resolución:
1. Establécese como límites máximos de concentración de nitrosaminas (en ppm) permitidos en los productos que contienen el principio activo ranitidina, los indicados en la Tabla Nº 2, aplicando la ecuación:
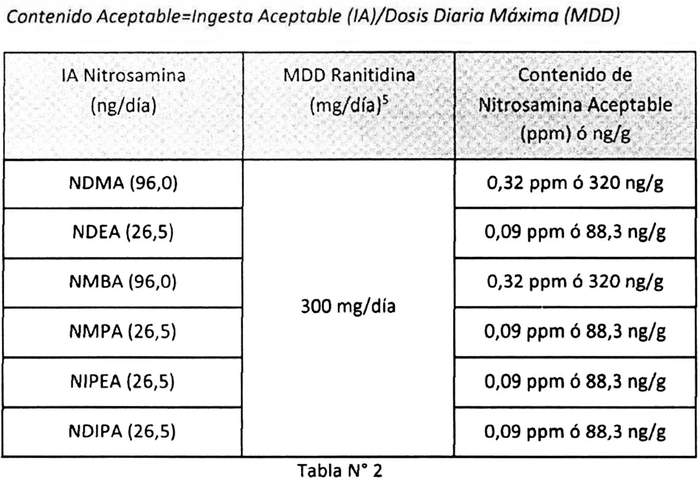
------------------------
(5) Ranitidine. Clinical Pharmacology [Internet] Tampa (FL): Elsevier. c2017- [citado 28 septiembre 2020]. Disponible en: http://www.clinicalpharmacology.com.
2. Incorpórase como análisis de carácter obligatorio, la determinación y cuantificación de las impurezas de nitrosaminas (Tabla Nº 2) a las especificaciones de producto terminado de los registros sanitarios de productos que contienen el principio activo ranitidina, presentando la respectiva modificación de especificaciones de producto terminado a través de la prestación código arancel 4111020 (Modificación de Especificaciones de Producto Farmacéutico o Pesticida por Producto).
3. Instrúyese a los titulares de registros sanitarios de productos que contienen el principio activo ranitidina, la actualización de la metodología analítica (farmacopea reconocida), mediante la cual se identificarán y cuantificarán las impurezas nitrosaminas (Tabla Nº 2) y presentarla al Instituto a través de la prestación código arancel 4111019 (Modificación de Metodología Analítica Producto Farmacéutico).
4. Instrúyese a los titulares de registros sanitarios de productos que contienen el principio activo ranitidina, la realización de estudios de estabilidad en tiempo real, incorporando la determinación de impurezas de nitrosaminas (tabla Nº 2), de acuerdo a la metodología validada mencionada en los puntos 1 y 2, y la presentación de sus resultados a este Instituto por todo el período, considerando que las cantidades no pueden exceder el límite establecido en el punto 1.
5. Déjase establecido que, para autorizar el Uso y Disposición de productos que contienen ranitidina, el solicitante deberá acreditar, para cada lote, mediante los certificados de análisis correspondientes que la cantidad de impurezas detectada es inferior al límite máximo definido en la Tabla Nº 2 y que la cuantificación de impurezas detectada es inferior al límite máximo definido en la Tabla Nº 2 y que la cuantificación de impurezas nitrosaminas fue realizada mediante el método autorizado por este Instituto en el punto 3 para productos registrados.
6. Déjase establecido que el plazo de presentación de las modificaciones de especificaciones de producto terminado y de metodología analítica es de 30 días a partir de la fecha de publicación de la presente resolución.
7. Previénese que el artículo 174 del Código Sanitario dispone que: "La infracción de cualquiera de las disposiciones de este Código o de sus reglamentos y de las resoluciones que dicten los Directores de los Servicios de Salud o el Director del Instituto de Salud Pública de Chile, según sea el caso, salvo las disposiciones que tengan una sanción especial, será castigada con multa de un décimo de unidad tributaria mensual hasta mil unidades tributarias mensuales. Las reincidencias podrán ser sancionadas hasta con el doble de la multa original" (énfasis agregado).
8. Déjase establecido que los límites para las especificaciones de producto terminado, la metodología analítica validada y el estudio de estabilidad que incluya el control de las nitrosaminas son requisitos para la obtención de nuevos registros de productos farmacéuticos orales que incluyan ranitidina.
9. Déjase establecido que las medidas dispuestas no interfieren, ni invalidan otras acciones de carácter sanitario que la autoridad determine.
Anótese, comuníquese y publíquese en el Diario Oficial.- Heriberto García Escorza, Director (S), Instituto de Salud Pública de Chile.